

Abstract
MAb1, a human IgG1 monoclonal antibody produced in a NS0 cell line, exhibits charge heterogeneity because of the presence of variants formed by processes such as N-terminal glutamate cyclization, C-terminal lysine truncation, deamidation, aspartate isomerization and sialylation in the carbohydrate moiety. Four major charge variants of MAb1 were isolated and the conformations of these charge variants were studied using hydrogen/deuterium exchange mass spectrometry, including the H/D exchange time course (HX-MS) and the stability of unpurified proteins from rates of H/D exchange (SUPREX) techniques. HX-MS was used to evaluate the conformation and solution dynamics of MAb1 charge variants by measuring their deuterium buildup over time at the peptide level. The SUPREX technique evaluated the unfolding profile and relative stability of the charge variants by measuring the exchange properties of globally protected amide protons in the presence of a chemical denaturant. The H/D exchange profiles from both techniques were compared among the four charge variants of MAb1. The two techniques together offered extensive understanding about the local and subglobal/global unfolding of the charge variants of MAb1. Our results demonstrated that all four charge variants of MAb1 were not significantly different in conformation, solution dynamics and chemical denaturant-induced unfolding profile and stability, which aids in understanding the biofunctions of the molecules. The analytical strategy used for conformational characterization may also be applicable to comparability studies done for antibody therapeutics.
Keywords: monoclonal antibody (mAb), IgG1, charge heterogeneity, hydrogen/deuterium exchange mass spectrometry, ion exchange chromatography, protein conformation, folding/unfolding stability
Introduction
The development of monoclonal antibody (mAb) therapeutics has become the fastest growing segment in the biopharmaceutical industry, with applications mainly in the treatment of immunological, oncological and inflammatory diseases.1 During production or storage, mAbs are subjected to post-translational and chemical modifications, such as glycosylation, C-terminal lysine truncation, N-terminal glutamine cyclization, deamidation, sialylation, oxidation, aspartate isomerization, hydrolysis and aggregation.2,3 These modifications may have substantial effects on the quality, safety and efficacy of the products. Some of these modifications can contribute to the heterogeneity of purified mAbs, for example, by altering their surface charge properties. The charge related heterogeneity of the mAbs can be resolved through ion exchange chromatography (IEC) or other charge-based separation methods, e.g., isoelectric focusing (IEF).4,5
Charge variants of a recombinant humanized IgG1 were previously isolated at Genentech using cation exchange displacement chromatography and reported to have similar in vitro potency or in vivo pharmacokinetics (PK) in rats.3 The marginal changes in FcRn binding affinity of the isolated acidic charge variant did not produce major changes in the potency or PK. Circular dichroism characterization performed with the charge variants revealed no major secondary and tertiary structure differences. Although there are no global changes in the structure, it is possible that secondary and tertiary structure may differ at some local regions of these charge variants. Technologies such as H/D exchange mass spectrometry (MS) can be effectively used to monitor the local structural changes of proteins.6-8
In this study, we used two H/D exchange MS techniques, the H/D exchange MS time course method (HX-MS)6,8 and stability of unpurified proteins from rates of H/D exchange (SUPREX),9 to study the charge variants of another antibody, MAb1, which is a human IgG1 produced in a NS0 cell line. In both techniques, pepsin digestion was used to digest the proteins so that structural information at the peptide or domain level can be obtained. The HX-MS approach measures deuterium buildup in a protein over time courses and provides information regarding conformation and solution dynamics of the protein.8 HX-MS has been successfully applied to conformation analysis of IgG1 for both unmodified and modified forms produced through deglycosylation or oxidation.6,7,10 SUPREX, in contrast to the HX-MS method where the backbone amide protons exchange when proteins locally unfold, uses a chemical denaturant to induce unfolding reactions of proteins and measures the exchange of backbone amide protons that exchange only when proteins subglobally or globally unfold.11-13 This technique can be used to investigate unfolding and measure unfolding stability of a protein either qualitatively or quantitatively.9,12,14 With the use of enzymatic digestion, SUPREX is also able to probe the unfolding stability of a specific domain of a multi-domain protein.15 SUPREX is well suited to be a subclass of various H/D exchange mass spectrometry techniques since it uses H/D exchange and mass spectrometry readout. SUPREX was originally developed to study the stability of unpurified proteins using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) without separation of the peptides. Most of the reported SUPREX data were acquired by MALDI-MS.12,15 The SUPREX experiments described here used electrospray ionization mass spectrometry (ESI-MS) to acquire mass spectra of the peptic fragments of MAb1 after reversed-phase separation.
The isolated charge variants of MAb1 include one acidic form and three other charge variants associated with C-terminal lysine clipping. Our aims were to identify the conformational differences among the charge variants of MAb1 in their localized regions (at peptide level), compare the unfolding profile and relative unfolding stability of these samples at their domain level and provide structural level evidence to correlate C-terminal Lys truncation of IgG1 to receptor binding activity.
Results
Cation exchange chromatography (CEX) separation of MAb1 and characterization of the charge variants
MAb1 can be resolved into four distinct peaks when subjected to CEX chromatography separation (Fig. 1). The most abundant peak (main peak) is termed K0 and the two basic peaks are termed K1 and K2, respectively. The peptide mapping data of the charge variants in addition to masses of the reduced samples and carboxypeptidase B treatment results confirmed that K0 has all C-terminal Lys truncated, K1 with one C-terminal Lys truncated in one of the heavy chains and K2 with no C-terminal Lys truncation (Table 1). It was also noted that the acidic peak consists of mainly C-terminal Lys truncated form of the antibody with higher levels of deamidated, sialylated and oxidized forms than the main peak, K0 (Table 1). Apart from the C-terminal Lys composition, the K0, K1 and K2 differ slightly in their oxidation levels (Table 1).

Figure 1. Representative cation exchange chromatography profile of MAb1.
Table 1. Analytical characterization of the isolated charge variants of MAb1.
| Modifications | Acidic peak | K0 | K1 | K2 |
|---|---|---|---|---|
| HC C-terminal Lys variants |
1.0% |
0.6% |
42.8% |
92.7% |
| Deamidation at Fc N388 |
19.6% |
1.6% |
1.2% |
1.8% |
| Sialylation |
14.5% |
1.3% |
0.5% |
0.3% |
| Oxidation at M256 |
6.7% |
3.2% |
5.0% |
7.7% |
| Oxidation at M432 | 3.5% | 1.2% | 2.4% | 4.8% |
The acidic peak, K0, K1 and K2 constitute approximately 13%, 60%, 10% and 8% of the intact MAb1, respectively, in terms of the integrated peak area percentage (Fig. 1). The CEX was used to fractionate MAb1 and generate milligram quantities for each of the four charge variants. The fractions of each charge variants were concentrated and buffer exchanged to PBS buffer. The concentrated samples were re-injected for CEX analysis to confirm their identity (data not shown). The purity was estimated to be greater than 95% for all isolated charge variants based on the integrated CEX peak area.
Biacore binding assay
The binding of these charge variants to the innate receptor of MAb1 was measured using Biacore along with the starting material of MAb1. The binding activities of these charge variants to receptor are 98%, 109%, 99% and 93% for the acidic peak, K0, K1 and K2, respectively, indicating that they are not significantly different from each other and are comparable to the starting MAb1 (90%), and that the C-terminal Lys truncation did not affect binding activity of the molecule.
HX-MS analysis
The HX-MS method was used to measure the exchange of locally protected backbone amide protons of MAb1 charge variants over time courses. The deuteration level of each charge variant of MAb1 was measured at ambient temperature. Figure 2 shows the deuteration level of MAb1 charge variant K0 (the main peak) for the light chain (LC) and heavy chain (HC). For each peptide, the deuteration levels at four time points (60, 300, 3,000 and 10,000 sec) are shown in color codes. Except for a few regions that exchanged fast (up to 70–90%) in the LC, the majority of the LC and HC exchanged at moderate or low levels (10–50%) under our experimental conditions.

Figure 2. Deuteration level in color scheme for the light chain (A) and heavy chain (B) of MAb1 charge variant K0. Each horizontal block represents a peptide (the two N-terminal amino acid residues were truncated). Each vertical block represents an exchange time, from top to bottom for 60, 300, 3,000 and 10,000 sec. The color codes were shown at the bottom. The labeled secondary structure as shown in β-sheets and other symbols were based on the X-ray crystallography structures of the Fab of MAb1 and Fc (3AVE.pdb) of IgG1. The CDR regions were highlighted by orange bars above the secondary structure symbols.
The deuteration levels of K0 at 10,000 sec were imposed onto the X-ray crystallography structures of both Fab (data undisclosed) and Fc (3AVE.pdb) using PyMOL (The PyMOL Molecular Graphics System, Schrödinger, LLC)(Fig. 3). This offered a different view of the data to compare which region exchanged fast or slow at 10,000 sec and which regions are covered by our data. As shown in Figures 2 and 3, the sequence coverage was ~85% for the Fab region and ~50% for the Fc region of the MAb1 K0.

Figure 3. Deuteration level of MAb1 charge variant K0 at 10,000 sec modeled to the X-ray crystallography structures, (A) for Fab region of MAb1 and (B) for Fc region. HX-MS data are not available for the grayed-out regions. Each color other than gray represents a deuteration level of a specific peptide. The color codes were shown at the bottom. Both structures were prepared using PyMOL.
The H/D exchange data from the HX-MS experiments for all charge variants are compiled in Table 2. After 60 sec exchange, the deuteration level of K0 varied from 13% to 42% for the covered regions in HC and 13% to 63% for LC regions. The deuteration levels gradually increased at longer exchange times and reached a maximum of 95% at 10,000 sec. For the C-terminal Lys variants, K0, K1 and K2, the HC C-terminal peptides consisting of residues 430–450 or 430–451 showed similar deuteration levels across the tested time scale (Table 2). Comparison of the deuteration levels of the acidic peak, K1 and K2 to those of the K0 indicated differences in H/D exchange level ranging from -4% to 7%. This range is within the experimental error that is typically associated with HX-MS experiments.16,17
Table 2. Deuteration level differences between the MAb1 charge variant K0 (main peak) and other three charge variants at peptide level.
| MAb1 charge variant K0 | MAb1 acidic peak vs. K0 | MAb1 K1 vs. K0 | MAb1 K2 vs. K0 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start |
End |
Charge |
60 sec |
300 sec |
3000 sec |
10000 sec |
60 sec |
300 sec |
3000 sec |
10000 sec |
60 sec |
300 sec |
3000 sec |
10000 sec |
60 sec |
300 sec |
3000 sec |
10000 sec |
| HC7 |
20 |
2 |
34% |
39% |
54% |
54% |
2% |
1% |
1% |
0% |
0% |
-1% |
3% |
0% |
1% |
1% |
2% |
1% |
| 22 |
37 |
3 |
13% |
13% |
17% |
17% |
0% |
0% |
-1% |
1% |
-1% |
-1% |
-2% |
-2% |
-3% |
0% |
-1% |
1% |
| 41 |
52 |
2 |
20% |
25% |
31% |
32% |
0% |
-1% |
0% |
-1% |
0% |
0% |
2% |
1% |
0% |
1% |
1% |
1% |
| 55 |
71 |
2 |
42% |
48% |
61% |
59% |
0% |
0% |
0% |
-1% |
1% |
-1% |
3% |
-1% |
1% |
2% |
2% |
1% |
| 74 |
94 |
2 |
24% |
25% |
28% |
28% |
1% |
0% |
1% |
0% |
0% |
0% |
2% |
1% |
2% |
2% |
1% |
1% |
| 106 |
116 |
2 |
17% |
26% |
42% |
43% |
2% |
1% |
1% |
-1% |
2% |
2% |
5% |
0% |
3% |
2% |
1% |
1% |
| 110 |
116 |
1 |
18% |
24% |
34% |
40% |
0% |
-1% |
3% |
2% |
1% |
2% |
1% |
0% |
2% |
2% |
1% |
3% |
| 119 |
148 |
3 |
31% |
31% |
37% |
41% |
0% |
1% |
1% |
2% |
0% |
0% |
1% |
3% |
-1% |
2% |
2% |
4% |
| 119 |
149 |
3 |
27% |
28% |
33% |
37% |
0% |
-1% |
0% |
0% |
0% |
0% |
0% |
1% |
0% |
0% |
0% |
1% |
| 151 |
178 |
3 |
30% |
34% |
41% |
42% |
0% |
0% |
-1% |
1% |
0% |
-1% |
0% |
-1% |
1% |
0% |
0% |
0% |
| 152 |
178 |
3 |
32% |
38% |
44% |
46% |
0% |
-1% |
0% |
1% |
0% |
0% |
0% |
0% |
0% |
1% |
1% |
1% |
| 162 |
178 |
2 |
39% |
45% |
50% |
49% |
1% |
0% |
0% |
1% |
1% |
1% |
2% |
2% |
1% |
2% |
3% |
4% |
| 186 |
201 |
1 |
38% |
38% |
38% |
42% |
0% |
0% |
-2% |
1% |
1% |
1% |
-1% |
2% |
1% |
-1% |
2% |
2% |
| 203 |
225 |
3 |
32% |
36% |
37% |
41% |
-1% |
-1% |
-1% |
2% |
-1% |
2% |
-1% |
0% |
-1% |
0% |
0% |
1% |
| 241 |
245 |
1 |
27% |
34% |
58% |
57% |
2% |
1% |
1% |
3% |
4% |
1% |
6% |
2% |
4% |
5% |
4% |
4% |
| 247 |
256 |
2 |
20% |
28% |
52% |
59% |
-2% |
1% |
-1% |
1% |
1% |
2% |
0% |
2% |
0% |
5% |
4% |
3% |
| 259 |
266 |
1 |
28% |
27% |
29% |
33% |
1% |
-4% |
-3% |
4% |
3% |
-1% |
-2% |
2% |
0% |
-5% |
-2% |
1% |
| 313 |
335 |
3 |
14% |
18% |
26% |
32% |
0% |
3% |
-2% |
-3% |
0% |
0% |
-1% |
-1% |
2% |
2% |
5% |
1% |
| 375 |
380 |
1 |
24% |
27% |
39% |
44% |
2% |
-3% |
1% |
0% |
2% |
-2% |
1% |
3% |
4% |
-2% |
3% |
1% |
| 375 |
384 |
2 |
23% |
26% |
36% |
42% |
-1% |
1% |
-2% |
0% |
1% |
1% |
0% |
1% |
1% |
2% |
0% |
2% |
| 375 |
402 |
3 |
25% |
31% |
36% |
38% |
-2% |
1% |
2% |
2% |
-1% |
1% |
0% |
0% |
-1% |
-1% |
0% |
1% |
| 386 |
402 |
2 |
33% |
41% |
48% |
48% |
1% |
1% |
0% |
3% |
0% |
0% |
2% |
2% |
2% |
2% |
2% |
4% |
| 387 |
408 |
2 |
31% |
35% |
38% |
38% |
1% |
0% |
0% |
1% |
4% |
1% |
1% |
1% |
3% |
1% |
0% |
1% |
| 411 |
427 |
3 |
27% |
31% |
38% |
38% |
-2% |
0% |
0% |
-1% |
-1% |
1% |
-1% |
-3% |
-1% |
2% |
0% |
-1% |
| 417 |
429 |
2 |
32% |
38% |
46% |
49% |
1% |
1% |
0% |
-1% |
1% |
0% |
0% |
1% |
3% |
0% |
3% |
3% |
| 430 |
450 |
3 |
25% |
28% |
33% |
38% |
2% |
0% |
0% |
0% |
0% |
-1% |
0% |
-1% |
NA |
NA |
NA |
NA |
| 430 |
451* |
3 |
NA |
NA |
NA |
NA |
NA |
NA |
NA |
NA |
25% |
28% |
36% |
39% |
25% |
30% |
38% |
40% |
| LC3 |
11 |
1 |
55% |
66% |
73% |
74% |
0% |
2% |
-3% |
1% |
1% |
3% |
-2% |
-2% |
4% |
1% |
-2% |
0% |
| 3 |
21 |
2 |
39% |
45% |
53% |
57% |
0% |
4% |
2% |
1% |
1% |
0% |
2% |
0% |
1% |
1% |
1% |
-2% |
| 3 |
32 |
3 |
34% |
40% |
52% |
56% |
0% |
1% |
0% |
0% |
0% |
0% |
0% |
-1% |
-1% |
0% |
1% |
2% |
| 14 |
23 |
1 |
30% |
40% |
52% |
54% |
-4% |
3% |
-4% |
5% |
-3% |
0% |
4% |
6% |
-5% |
-5% |
-1% |
0% |
| 34 |
46 |
3 |
30% |
33% |
38% |
38% |
-1% |
1% |
2% |
0% |
0% |
-1% |
1% |
-1% |
1% |
-1% |
1% |
0% |
| 35 |
46 |
2 |
28% |
31% |
38% |
40% |
0% |
1% |
0% |
1% |
0% |
0% |
1% |
1% |
1% |
1% |
2% |
0% |
| 40 |
52 |
2 |
18% |
20% |
26% |
28% |
0% |
1% |
0% |
1% |
1% |
0% |
1% |
1% |
1% |
1% |
1% |
0% |
| 49 |
62 |
2 |
35% |
34% |
44% |
47% |
1% |
4% |
1% |
1% |
1% |
-1% |
1% |
2% |
-2% |
-1% |
3% |
1% |
| 51 |
71 |
3 |
36% |
39% |
44% |
46% |
-3% |
0% |
-2% |
1% |
-2% |
2% |
0% |
0% |
-2% |
2% |
0% |
2% |
| 73 |
82 |
1 |
13% |
18% |
29% |
32% |
0% |
0% |
0% |
1% |
1% |
0% |
2% |
1% |
1% |
1% |
2% |
1% |
| 77 |
83 |
1 |
28% |
35% |
46% |
48% |
-2% |
-1% |
0% |
0% |
0% |
0% |
2% |
0% |
1% |
2% |
1% |
2% |
| 89 |
116 |
3 |
23% |
24% |
28% |
31% |
2% |
1% |
0% |
1% |
0% |
0% |
-1% |
1% |
1% |
0% |
1% |
1% |
| 91 |
120 |
3 |
20% |
21% |
25% |
29% |
2% |
0% |
1% |
-1% |
1% |
0% |
0% |
-1% |
2% |
0% |
0% |
0% |
| 94 |
109 |
3 |
14% |
18% |
20% |
21% |
-3% |
-1% |
2% |
1% |
-2% |
1% |
1% |
0% |
-2% |
-1% |
1% |
1% |
| 118 |
122 |
1 |
25% |
39% |
43% |
38% |
4% |
2% |
0% |
1% |
0% |
6% |
7% |
0% |
3% |
5% |
5% |
3% |
| 118 |
132 |
2 |
42% |
47% |
49% |
52% |
1% |
0% |
0% |
-1% |
2% |
2% |
1% |
1% |
2% |
1% |
2% |
1% |
| 119 |
132 |
2 |
50% |
55% |
57% |
62% |
2% |
0% |
-1% |
1% |
2% |
1% |
0% |
3% |
2% |
1% |
-1% |
2% |
| 139 |
150 |
2 |
63% |
73% |
93% |
95% |
-1% |
0% |
-2% |
0% |
-4% |
-2% |
5% |
2% |
-5% |
-1% |
2% |
4% |
| 164 |
179 |
2 |
16% |
19% |
24% |
27% |
1% |
1% |
-3% |
1% |
1% |
1% |
0% |
0% |
1% |
3% |
1% |
2% |
| 183 |
196 |
3 |
17% |
13% |
19% |
20% |
-2% |
1% |
-1% |
0% |
1% |
-3% |
-3% |
-3% |
-4% |
-1% |
-1% |
-1% |
| 199 | 213 | 2 | 32% | 32% | 41% | 48% | 3% | 2% | 2% | 3% | 2% | -3% | -1% | 2% | 1% | 2% | 5% | 5% |
*The percentage values listed for peptide HC 430–451 under "K1 vs. K0" or "K2 vs. K0" columns are the deuteration levels for K1 or K2, respectively.
SUPREX analysis
SUPREX is a technique analogous to conventional denaturation-induced equilibrium assay for unfolding stability measurement of a protein by using a chemical denaturant, such as GuHCl, to induce folding/unfolding equilibrium. In this assay, the C1/2SUPREX, i.e., the concentration of denaturant where 50% of globally protected amide protons in a protein is exchanged, can be used as a qualitative measure of the unfolding stability of a protein vs. its modified forms.12 In addition to this C1/2SUPREX parameter, the plot of deuteration level of a protein or peptide as a function of denaturant concentration can also be used to compare the unfolding behavior of a protein vs. its modified forms.12 Therefore, SUPREX is well-suited to compare the unfolding profile and unfolding stability among the charge variants of MAb1.
In SUPREX, high sequence coverage may not be necessary; however, results for representative analogous peptides from each individual domain of the tested protein are helpful to obtain a better assessment of the unfolding behavior and unfolding stability of the protein.15 The regions of K0 for which SUPREX data were generated are shown in Figure 4 (labeled in non-gray colors; those without SUPREX data were grayed out). Ten peptides, five from the LC and five from the HC were selected for analysis because of their high signal to noise ratios. These peptides are either from the variable and constant regions of the LC or from the viable region, CH1, CH2 and CH3 domains of the HC (Table 3). The deuteration profiles of these peptides in SUPREX were determined for all four charge variants.

Figure 4. Regions of MAb1 charge variant K0 covered by SUPREX data. Each color other than gray represents a peptide for which SUPREX data are available. SUPREX data are not available for the grayed-out regions.
Table 3. C1/2SUPREX values for MAb1 charge variants.
| Peptides | Regions | Charge | Acidic peak (M GuHCl) |
K0 (M GuHCl) |
K1 (M GuHCl) |
K2 (M GuHCl) |
|---|---|---|---|---|---|---|
| HC 53–65 |
HC Variable |
1 |
N/A |
N/A |
N/A |
N/A |
| HC 117–148 |
HC CH1 |
2 |
3.6 |
3.6 |
3.6 |
3.6 |
| HC 150–178 |
HC CH1 |
3 |
3.7 |
3.6 |
3.6 |
3.6 |
| |
|
|
3.7* |
3.6* |
3.6* |
3.6* |
| HC 246–256 |
HC CH2 |
1 |
N/A |
N/A |
N/A |
N/A |
| HC 381–408 |
HC CH3 |
2 |
3.9 |
4.1 |
4.1 |
3.9 |
| |
|
|
|
|
|
|
| LC 1–32 |
LC Variable |
3 |
3.6 |
3.7 |
3.7 |
3.6 |
| LC 33–46 |
LC Variable |
2 |
3.5 |
3.6 |
3.5 |
3.6 |
| LC 47–70 |
LC Variable |
2 |
3.5 |
3.5 |
3.6 |
3.5 |
| LC 71–82 |
LC Variable |
1 |
3.6 |
3.5 |
3.5 |
3.5 |
| |
|
|
3.6** |
3.6** |
3.6** |
3.6** |
| LC 116–132 | LC constant | 2 | 3.5 | 3.6 | 3.6 | 3.6 |
Average C1/2 SUPREX value for HC CH1 region from HC 117–148 and 150–178. **Average C1/2 SUPREX value for LC variable region from LC 1–32, 33–46, 47–70 and 71–82. Experimental error for SURPEX measurement is typically ± 0.1 M GuHCl.
Representative SUPREX deuteration plots for the four charge variants of MAb1 are shown in Figure 5. HC 53–65, 117–148, 246–256 and 381–408 are from the HC variable region, CH1, CH2 and CH3 domains, respectively, of MAb1. LC 33–46 and 116–132 are representative peptides from the variable and constant region, respectively, of MAb1 LC. The deuteration level plots for these peptides are visually similar among all four charge variants, suggesting the unfolding profiles of these charge variants in these regions are not significantly different. The HC 53–65 and 246–256 peptides showed almost full deuteration over the range of the GuHCl concentrations, implying no globally or subglobally protected amide protons in these regions during the 30 min exchange period. SUPREX-like (sigmoidal) curves can be fit for some peptides from the four charge variants as shown in Figure 5 for HC 117–148, HC 381–408, LC 33–46 and LC 116–132. For HC CH3 domain peptide 381–408, the acidic peak seemed to show slightly higher deuteration levels in the pre-transition region than the other three charge variants. The differences (< 0.5 Da), however, are not significant and did not translate to a measurable difference in the transition midpoint of the SUPREX curve (the C1/2SUPREX) (Table 3). The C1/2SUPREX values extracted for all the analyzed peptides of the charge variants except HC 53–65 and HC 246–256 are tabulated in Table 3. The C1/2SUPREX values range from 3.5 M to 3.7 M GuHCl for the Fab peptides from either the HC or LC, and 3.9 - 4.1 M GuHCl for an Fc peptide (HC CH3 domain). In other words, peptides from the Fab region of either the HC or LC showed similar C1/2SUPREX values, and the peptide from the CH3 domain showed a slightly higher C1/2SUPREX value, than those of the peptides from the Fab regions.

Figure 5. SUPREX deuteration plots for representative peptides of MAb1 charge variants. Acidic peak (circles filled in black, black solid line); K0 (open circles, long dash line), K1 (circles filled in red, red line) and K2 (diamonds, blue line).
DSC was performed for the MAb1 starting material and its four charge variants to compare their thermal unfolding stability. DSC was also performed on the isolated (Fab)2, Fab and Fc fragments of MAb1 for domain assignment. The thermal unfolding curve of full-length MAb1 by DSC showed two distinct transitions with melting temperature (Tm) at ~70°C for the small-amplitude transition and ~80°C for the large-amplitude transition, typical for humanized IgG1 as reported by Ionescu.18 The four charge variants of MAb1 showed similar thermal denaturation profiles to each other and also to the starting material (data not shown). The Tm values for both transitions are also similar among them (Table 4). The isolated (Fab)2 and Fab present only one peak around 80°C that align with the large-amplitude transition of MAb1. In comparison, the isolated Fc of MAb1 showed two peaks with denaturation profile similar to those of the IgG1 Fc fragments reported previously.18-20 According to these reports, the first transition of Fc should correspond to the unfolding of the CH2 domain, and the second transition is related to the CH3 domain. The Tm of the CH3 domain of MAb1 Fc fragment is 0.4°C greater than the Tm2 of the intact MAb1 and Tm of the isolated Fab or (Fab)2; however, the Tm of the CH2 domain of Fc fragment is 1.1°C lower than the Tm1 of the intact MAb1 (Table 4). Overall, the DSC results of (Fab)2, Fab and Fc of MAb1 confirmed that the first Tm of MAb1 is associated with the CH2 domain unfolding, and the second Tm is Fab and CH3 domain associated.
Table 4. DSC data of MAb1 charge variants and fragments.
| Samples | Tm1 (°C) | Tm2 (°C) |
|---|---|---|
| MAb1 Starting Material |
71.6 |
84.4 |
| MAb1 (Fab)2 |
NA |
84.4 |
| MAb1 Fab |
NA |
84.4 |
| MAb1 Fc |
70.5 |
84.8 |
| MAb1 Acidic Peak |
71.7 |
84.5 |
| MAb1 K0 |
71.8 |
84.6 |
| MAb1 K1 |
71.4 |
84.4 |
| MAb1 K2 | 71.4 | 84.2 |
Note: Experimental error for DSC measurement is typically < 0.2°C.
Discussion
The H/D exchange profiles of four charge variants of MAb1, i.e., acidic form, K0, K1 and K2 were studied using two H/D exchange mass spectrometry techniques for biophysical characterization, including conformation and dynamics in solution, unfolding profile and stability. The deuteration levels of MAb1 K0 measured in HX-MS experiments (Fig. 2; Table 2) indicated that most of the regions of MAb1 K0 exchanged at moderate level, up to 50% after 10,000 sec exchange. It is interesting to note that one of the HC variable regions of peptide 22–37, which spans the CDR1 region of MAb1, exchanged minimally (~10%) over the tested time scale. The low exchange rates of this region may be attributed to its low dynamics in solution, hydrogen bonding or low solvent accessibility of amide protons. In comparison, Burkitt et al. also reported very slow exchange for some variable regions of the HC of another IgG1 antibody.10 It appears that in reality some variable regions of the IgG1 may be highly protected and have low solution dynamics.
The sequence coverage by the HX-MS data is high for the Fab regions of MAb1 (up to 85%) and low for the Fc region (~50%). The limited sequence coverage for the Fc region of MAb1 was likely attributed to the glycosylation of Asn301 and inefficient reduction of the S-S bonds under the quenching and digestion conditions. Slightly longer digestion time in ice, with a more efficient quench buffer or digestion at room temperature may help to improve the Fc sequence coverage. No significant deuteration level differences were detected over the four time points between K0 and other three charge variants for either the HC or LC in the covered regions of MAb1 (Table 2), suggesting that the conformation and solution dynamics of these charge variants are similar.
In SUPREX, the deuteration plot of a protein or peptide as a function of a chemical denaturant may show different profiles. One scenario is the mass increase of the protein or peptide is approximately the same over the entire range of chemical denaturant concentrations used in the study, e.g., plots shown in Figure 5 for peptide HC 53–65 and 246–256, which indicates that exchanges of the backbone amide protons are not dependent on the chemical denaturant. All backbone amide protons were exchanged during the exchange time scale even in the absence of a chemical denaturant. The data for these two peptides demonstrated that the unfolding profiles of these regions are similar among the four charge variants; however, they did not provide conclusive information regarding the unfolding stability of the corresponding regions. It is likely that these two regions were not as stable as other regions from which SUPREX transition midpoints were extracted. The other possibility is that these two peptides may not represent the unfolding stability of the HC variable region (by HC 53–65) or the CH2 domain (by HC 246–256) of the HC. It is possible that the most stable peptides in these regions were not detected in our SUPREX tests. The use of high concentrations of GuHCl (up to 6.5M) in the SUPREX buffer or quench buffer may have limited the digestion efficiency of pepsin. As a consequence, less high quality peptides resulted in comparison to those in the HX-MS experiment. In addition, the shorter gradient (14 min) used in the separation of peptides in the SUPREX experiment may have also reduced resolution of the separation, therefore reducing the number of peptides that could be detected by MS. Longer gradients may increase the chance to resolve more peptides with high MS quality, to increase sequence coverage in SUPREX experiment and to offer better assessments of the unfolding stability of the regions as represented by HC 53–65 and HC 246–256.
Another scenario that is typical in the SUPREX test occurs when the deuteration plot looks like a sigmoidal curve, e.g., the curves shown in Figure 5 for HC 117–148, 381–408, LC 33–46 and 116–132. The mass increase in the pre-transition region of the curve is dependent on the number of amide protons that exchange when a protein or peptide locally unfolds. In the post-transition region, all amide protons in a protein or peptide are exchanged. The amplitude between the pre-transition and post-transition lines of the SUPREX curve corresponds to the number of globally or subglobally protected amide protons that exchange only when proteins or peptides globally or subglobally unfold.
C1/2SUPREX values were extracted for HC 117–148, HC 381–408, LC 33–46, LC 116–132, as well as other peptides from MAb1, and used as a qualitative measure of the unfolding stability of their representative domains (Fig. 5; Table 3). In our case, this parameter was especially useful for comparing the unfolding stability of all charge variants of MAb1. The SUPREX results suggested a minimum of two distinct unfolding transitions according to the two groups of C1/2SUPREX values, one for the Fab regions and the other for the Fc region represented by HC 381–408, the CH3 domain (Table 3). The similar C1/2SUPREX values among the LC variable, LC constant and HC CH1 domains likely imply that these three domains unfold cooperatively. The consistently higher C1/2SUPREX value of the CH3 domain compared with that of the Fab domains does not necessarily mean that the CH3 domain is more stable than the Fab domains because the dependence of unfolding free energy as a function of [GuHCl] may be different between the Fab and CH3 domains. The determination of the dependence of unfolding free energy as a function of [GuHCl] for Fab and CH3 domains is out of the scope of this study. Nonetheless, the SUPREX data of the MAb1 charge variants suggest that the CH3 domain unfolds independently from the Fab domains.
There may be correlations between the transition midpoints obtained by SUPREX and melting temperatures (Tm) determined by DSC because both methods detect unfolding events. The SUPREX data demonstrate that the C1/2SUPREX values of the CH3 domains of MAb1 are up to 0.5 M GuHCl higher than those of the CH1/Fab domains of MAb1 (Table 3). Interestingly, the DSC data show that the Tm of the CH3 domain of the isolated Fc was slightly greater than that of the isolated Fab (84.8 vs. 84.4°C) (Table 4). However, such a Tm difference does not seem to be significant. It is worth noting that the Tm of the CH2 domain of isolated Fc is lower than that of the full-length MAb1 by 1.1°C. This suggests that the conformation of the isolated Fc slightly differs from when it is in the intact protein. With the resolution of DSC, the Tm of Fab and CH3 domain of Fc could not be unambiguously distinguished when they are intact in the MAb1. Overall, the DSC data helped to explain the SUPREX results about HC 246–256 from the CH2 domain of MAb1. This region did not show SUPREX transitions at 30 min exchange because it is not as stable as other regions from which SUPREX transition midpoints were extracted. The SUPREX derived unfolding stability data represented by the C1/2SUPREX values offered relatively higher resolution than DSC data because it covered the majority domains of the molecule, including the variable and constant regions of LC, the CH1 and CH3 domains of the HC.
The deuteration plots of all peptides of MAb1 in the SUPREX experiments showed that the exchange levels were close to each other among all four charge variants over the 12 GuHCl concentrations, providing evidence that the unfolding profiles induced by GuHCl are similar among these charge variants of MAb1. In summary, the SUPREX data suggest that all the tested charge variants of MAb1 are similar in their unfolding profile and unfolding stability.
C-terminal lysine variation is quite common for mAbs and was believed to be a quality attribute of a given mAb due to carboxypeptidase B-like activity in cell culture that may differ upon manufacturing changes, but not a critical quality attribute because the variation did not affect in vitro potency in complement dependent cytotoxicity or anti-proliferation, FcRn binding affinity or the pharmacokinetics properties in rats.3,5 The Biacore binding data of the three charge variants of MAb1 associated with C-terminal Lys processing confirmed that C-terminal Lys truncation did not affect binding of the molecule to the receptor. These facts were supported by our H/D exchange MS data demonstrating the similar conformations among the C-terminal Lys variants. The acidic charge variant of MAb1 showed greater modification differences from the other three C-terminal Lys variants, but its H/D exchange profiles are not significantly different from them. It is likely that the observed modifications of deamidation, sialylation and oxidation (Table 1) are not sufficient in inducing conformational changes in this acidic form of MAb1.
Different types of microheterogeneity can be introduced due to enzymatic processing or nonenzymatic modifications during the production of mAbs. The differences observed in the proportions of the charge variants of mAbs during routine manufacturing process changes can have a potential influence on the stability and biological activity of the target molecules, and they may pose a challenge for demonstration of product comparability. Hence, these variants need to be characterized to understand their effects on the stability, structure and function of mAb therapeutics. The H/D exchange MS data presented in this study provide extensive information regarding the local and subglobal/global unfolding of the charge variants of MAb1 at peptide or domain level resolution over a large sequence portion of MAb1. Our data indicate that there are no significant differences among the charge variants of MAb1 in conformation, solution dynamics, unfolding profile and stability. These results provide additional evidence to support the higher order structure similarity among the major charge variants of IgG1, and are therefore helpful for understanding the biofunctions of these charge variants. The analytical strategy employed in this study applies to the conformational characterization of the charge variants of mAbs and mAbs with post-translational, chemical or sequence modifications. It may also be applicable to many other instances where higher order structure characterization of protein therapeutics is needed, including comparability studies done after manufacturing changes and comparisons of biosimilar products to their reference molecules.
Materials and Methods
Materials
MAb1 was produced in NS0 cell lines and purified by the Process Development Group at ImClone Systems Corporation. Trypsin (sequencing grade TPCK modified) was obtained from Promega Corporation. Lyophilized porcine pepsin, sodium cyanoborohydride, 2M Na2SO4, 1M ethanolamine dithiothreitol (DTT), iodoacetamide, D2O, deuterated phosphoric acid, Tris(2-carboxyethyl)phosphine hydrochloride (TCEP•HCl) and formic acid were from Sigma-Aldrich. NAP-5 cartridges were from GE Healthcare. Guanidine hydrochloride (GuHCl), acetonitrile and TRIS-HCl buffer (pH 7.5) were from J. T. Baker. Sodium phosphate, sodium chloride and trifluoroacetic acid (TFA) were purchased from EMD Science or Fisher Scientific. POROS AL-20 resin was from Applied Biosystems.
Ion exchange chromatography
Cation exchange chromatography was performed with an analytical weak cation exchange column (Dionex ProPac WCX-10, 4.0 × 250 mm, 10 µm) using an Agilent 1100 series HPLC (Agilent Technologies). Gradient started from 100% buffer A (10 mM sodium phosphate, pH 7.2) to 22.5% B (200 mM NaCl in 10 mM sodium phosphate buffer, pH 7.2) in 90 min at a flow rate of 0.5 mL/min. The column temperature was kept at 50°C and detection was at 214 nm.
MAb1 was fractionated by CEX using the method described above. The four resolved charge variants were isolated and collected. All fractions were concentrated using Microcon filtration 10,000 MWCO (Millipore). Buffers of all concentrated fractions were exchanged to PBS (1X, GIBCO). The final concentration of each fraction was measured by UV spectrometer. The identity and purity of each concentrated fraction were confirmed by CEX. All fractions were diluted using PBS to 6.5 mg/ml before H/D exchange experiments.
Biacore binding assay
Biacore C (GE Healthcare) was used for binding activity measurement of the charge variants of MAb1. The innate antigen was immobilized onto a CM5 Sensor Chip using amine coupling method. A calibration curve was generated by injecting the MAb1 product standard that spans from 78–2,000 ng/mL onto the immobilized surface of the chip. Each charge variant was tested at five different concentrations from 158 ng/mL to 800 ng/mL based on the nominal protein concentration determined by UV spectrometer. The concentration of each charge variant was calculated from its resonance units (RU) by comparison to the RU of solutions containing known amounts of MAb1 standard, using a 4-parameter regression analysis. The potency values, reported as % binding activity, were calculated from mean concentration calculated for each charge variant from the five dilutions (determined by referring to the calibration curve) divided by the protein concentration measured by UV spectrometer and multiplied by 100.
Tryptic peptide mapping
200 µg of MAb1 charge variants were dried under vacuum. The reconstituted proteins were denatured and reduced by 7.2 M guanidine hydrochloride, 50 mM DTT in 100 mM Tris-HCl (pH 7.5) at 50°C for 60 min. The proteins were then alkylated with iodoacetamide for 30 min in the dark with constant shaking. The reaction mixture was then dialyzed against 50 mM Tris (pH 7.5) for 2 h using Slide-A-Lyzer Mini Dialysis Units (7000 MWCO, Pierce). The dialyzed solution was collected and digested by trypsin at 37°C for 3 h using an enzyme:protein ratio of 1:20 (w/w). The reaction was terminated by adding TFA to ~2%. The resultant peptides were resolved on a C18 reversed-phase column (Zorbax C18 300SB, 300 Å, 3.5 µm, 4.6 × 150 mm) at 50°C using an Agilent HPLC 1200 series interfaced to a LTQ ion trap mass spectrometer (Thermo Fisher Scientific) equipped with an electrospray ionization source and operated in a triple-play mode. The temperature of the heated capillary inlet was set at 300°C. The capillary voltage was set at 49 V, and the lens voltage was at 195V. The digested sample was eluted with a gradient from 99% solvent A (0.1% TFA) to 39% solvent B (100% acetonitrile in 0.085% TFA) in 95 min at a flow rate of 0.5 mL/min.
MAb1 (Fab)2, Fab and Fc fragments preparation
MAb1 was digested with immobilized pepsin (Pierce kit cat #44988) to generate (Fab)2 fragment, and was digested with immobilized papain (Pierce kit cat #44985) to generate Fab and Fc fragments following the manufacturer’s instructions. The (Fab)2, Fab and Fc fractions of MAb1 after Protein A purification were further purified by size-exclusion chromatography (SEC) using Agilent 1260 HPLC system and TSKGel G3000SWXL column (Tosoh Bioscience; 7.8 × 300 mm, 5 μm, 250Å). The mobile phase was 0.15 M NaCl in 10 mM sodium phosphate, pH 7.0, with a flow rate of 0.5 mL/min. All the purified (Fab)2, Fab and Fc fragments by SEC showed purity greater than 98.5%. The molecular weight of each purified fragment was measured using Waters Xevo G2 Q-TOF mass spectrometer to further confirm the identity of each fragment.
HX-MS experiment
A digestion condition optimization was performed to screen the best possible quench buffer that offered highest sequence coverage of MAb1 (non-fractionated) after digestion by pepsin. 5.8 M GuHCl in 0.8 M TCEP (pH 2.5) was used as the quench buffer. The concentrations of charge variants were 6.5 mg/ml in PBS buffer (pH 7.2). 7 µL of a 6.5 mg/ml MAb1 charge variant was mixed with 16.3 µL of a 50 mM sodium phosphate in D2O, pHread 7.2 to exchange for 60, 300, 3,000 and 10,000 sec in a water bath (20 ± 0.5°C). 35 µL of the quench buffer was added to quench the H/D exchange reaction. Quenched samples were frozen in dry ice and kept at -70°C. Frozen samples were defrosted at ambient temperature for 1 min and 30 sec, and injected into a pepsin column (packed in-house using an empty guard column (2.1 × 30 mm, Restek) and immobilized pepsin prepared as described elsewhere21). Pepsin digestion of the samples was performed in ice for 10 min. After digestion, the pepsin column was connected to an isocratic HPLC pump that delivered 600 µL of a 0.1% formic acid solution to transfer the resulting peptides onto a trap column (POROS 10 R2, Applied Biosystems) and also to desalt the samples. A switch valve (G1157A, Agilent Technologies) was set so that gradient from the binary pump of an Agilent 1100 HPLC eluted peptides from the peptide trap and a C18 column (Jupiter 1.0 × 50 mm, 5 µm, Phenomenex). The gradient was 90% buffer A (0.1% formic acid) to 40% buffer B (95% acetonitrile in 0.1% formic acid) over 28 min at a flow rate of 100 µL/min. All columns and connected tubing were immerged under ice to minimize back-exchange during reversed-phase separation. Mass spectrometric data were acquired with a Thermo Finnigan LCQ Deca XP mass spectrometer (Thermo Fisher) using the capillary temperature set at 225°C.
SUPREX experiment
Each charge variant of MAb1 was diluted to 6.5 mg/ml using PBS (1×) buffer, pH 7.2. The samples were exchanged for 30 min at ambient temperature in buffers containing 20 mM MOPS (pHread 7.0) and varied concentration of GuHCl. The GuHCl concentrations of the buffers ranged from 0.5 to 6.5 M with deuterium content of approximately 60%. The volume ratio of protein and the SUPREX buffer was approximately 1:3. Twenty-three point three micro liters of the exchanged samples were mixed with 35 µL of a quench buffer containing 5.8 M GuHCl/0.8 M TCEP (pH 2.5) and frozen in dry ice. The frozen samples were defrosted and injected to a pepsin column for a 10 min chill digestion. Peptides were desalted on a trap column and eluted from a C18 column with a gradient of 0.1% formic acid (A) and 95% acetonitrile in 0.1% formic acid (B) from 10% to 40% buffer B over 14 min. Data were acquired using Agilent 1100 HPLC instrument and LCQ Deca XP mass spectrometer.
DSC measurements
The DSC measurements were performed at 0.8 mg/mL protein and 200°C/hr scan rate from 10°C to 110°C on a VP-Capillary DSC Platform (MicroCal, LLC). All proteins were in PBS buffer, pH 7.2. The DSC profiles were calculated using the Origin 7.0 software. The buffer background and baseline were subtracted and the thermogram was normalized to the molar concentration of the protein. The melting temperature was then calculated for each peak in the thermogram by the software.
Data analysis
Data dependent acquisition MS/MS and SEQUEST were used for the identification of digested fragments of MAb1. Identity of each peptide was checked using SEQUEST score, MS isotopic pattern, retention time and molecular weight. A DXMS software (Sierra Analytics) was used to calculate the centroid mass value of each identified peptide with or without deuteration. Correction for back-exchange was not made for data reported here. Deuteration level of each peptide in the time course assay was calculated using Equation 1:22
 |
where m(P) and m(N) are the centroid value of partially deuterated and non-deuterated sample, respectively. MaxD is the maximum deuterium level calculated by subtracting two and the number of prolines in the third or later position of the peptide from the number of amino acids in the peptide of interest. This is based on the assumption that the first two amino acids of the derived peptides do not retain deuterium after RPLC separation.23,24 The reported sequence number of each peptide here corresponds to the full sequence of the peptide with truncation of the first two amino acids that are expected to carry no deuterium.
For SUPREX data analysis, the deuterium content of each peptide was calculated by subtracting the mass of the fully protonated sample from the average mass of the deuterated samples at each denaturant concentration. The deuterium contents (Da) of each peptide were plotted as a function of chemical denaturant concentration. The data were fitted to a 4-parameter sigmoidal equation (see below) using a nonlinear regression routine in SigmaPlot to extract the transition midpoint for each denaturation curve (i.e., the C1/2SUPREX value, the concentration of the denaturant at the transition midpoint of the sigmoidal curve where 50% of the globally protected amide protons were exchanged).
The C1/2SUPREX value was obtained by fitting the data using Equation 2:14
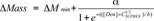 |
where ΔMmin is the mass change in the pre-transition baseline of the curve; a is the amplitude of the curve in Da; [Den] is the molar denaturant concentration; and b is a parameter that describes the steepness of the transition. The C1/2SUPREX value is used as a qualitative measure of the unfolding stability of the MAb1 charge variants.
Acknowledgment
We thank the Product Development Department at ImClone Systems for providing MAb1.
Disclosure of Potential of Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/22695
References
- 1.Sheridan C. Fresh from the biologic pipeline-2009. Nat Biotechnol. 2010;28:307–10. doi: 10.1038/nbt0410-307. [DOI] [PubMed] [Google Scholar]
- 2.Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr Pharm Biotechnol. 2008;9:468–81. doi: 10.2174/138920108786786402. [DOI] [PubMed] [Google Scholar]
- 3.Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs. 2010;2:613–24. doi: 10.4161/mabs.2.6.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick LW, Jr., Qiu D, Mahon D, Adamo M, Cheng KC. C-terminal lysine variants in fully human monoclonal antibodies: investigation of test methods and possible causes. Biotechnol Bioeng. 2008;100:1132–43. doi: 10.1002/bit.21855. [DOI] [PubMed] [Google Scholar]
- 5.Antes B, Amon S, Rizzi A, Wiederkum S, Kainer M, Szolar O, et al. Analysis of lysine clipping of a humanized Lewis-Y specific IgG antibody and its relation to Fc-mediated effector function. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;852:250–6. doi: 10.1016/j.jchromb.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 6.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2009;81:2644–51. doi: 10.1021/ac802575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics. 2010;9:1716–28. doi: 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–5. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghaemmaghami S, Fitzgerald MC, Oas TG. A quantitative, high-throughput screen for protein stability. Proc Natl Acad Sci USA. 2000;97:8296–301. doi: 10.1073/pnas.140111397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burkitt W, Domann P, O’Connor G. Conformational changes in oxidatively stressed monoclonal antibodies studied by hydrogen exchange mass spectrometry. Protein Sci. 2010;19:826–35. doi: 10.1002/pro.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/S0033583500005217. [DOI] [PubMed] [Google Scholar]
- 12.Fitzgerald MC, Tang L, Hopper ED. Thermodynamic analysis of protein folding and ligand binding by SUPREX. In: Barcelo, D, ed. Comprehensive Analytical Chemistry. Elsevier BV: Elsevier, 2009; 52: 127-49. [Google Scholar]
- 13.Maity H, Lim WK, Rumbley JN, Englander SW. Protein hydrogen exchange mechanism: local fluctuations. Protein Sci. 2003;12:153–60. doi: 10.1110/ps.0225803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powell KD, Fitzgerald MC. Accuracy and precision of a new H/D exchange- and mass spectrometry-based technique for measuring the thermodynamic properties of protein-peptide complexes. Biochemistry. 2003;42:4962–70. doi: 10.1021/bi034096w. [DOI] [PubMed] [Google Scholar]
- 15.Tang L, Roulhac PL, Fitzgerald MC. H/D exchange and mass spectrometry-based method for biophysical analysis of multidomain proteins at the domain level. Anal Chem. 2007;79:8728–39. doi: 10.1021/ac071380a. [DOI] [PubMed] [Google Scholar]
- 16.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, et al. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–92. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iacob RE, Engen JR. Hydrogen exchange mass spectrometry: are we out of the quicksand? J Am Soc Mass Spectrom. 2012;23:1003–10. doi: 10.1007/s13361-012-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 2008;97:1414–26. doi: 10.1002/jps.21104. [DOI] [PubMed] [Google Scholar]
- 19.Souillac PO. Biophysical characterization of insoluble aggregates of a multi-domain protein: an insight into the role of the various domains. J Pharm Sci. 2005;94:2069–83. doi: 10.1002/jps.20423. [DOI] [PubMed] [Google Scholar]
- 20.Ghirlando R, Lund J, Goodall M, Jefferis R. Glycosylation of human IgG-Fc: influences on structure revealed by differential scanning micro-calorimetry. Immunol Lett. 1999;68:47–52. doi: 10.1016/S0165-2478(99)00029-2. [DOI] [PubMed] [Google Scholar]
- 21.Busby SA, Chalmers MJ, Griffin PR. Improving digestion efficiency under H/D exchange conditions with activated pepsinogen columns. Int J Mass Spectrom. 2007;259:130–9. doi: 10.1016/j.ijms.2006.08.006. [DOI] [Google Scholar]
- 22.Zhang ZQ, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–31. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamuro Y, Coales SJ, Southern MR, Nemeth-Cawley JF, Stranz DD, Griffin PR. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J Biomol Tech. 2003;14:171–82. [PMC free article] [PubMed] [Google Scholar]
- 24.Bai YW, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]