

Abstract
C-reactive protein (CRP) is a serum marker highly upregulated in inflammation after bacterial infection. Robust, reliable and quick quantification of CRP would be a substitute for erythrocyte sedimentation rate (ESR) with superior diagnostic value. Quartz crystal microbalance (QCM) based sensors coated with specific antibodies and integrated into lab-on-chip systems are in development for rapid point of care quantification. In this study, we isolated three CRP specific single chain (sc)Fv antibody fragments using phage display from an antibody gene library. Their affinities ranged from 2.7 × 10−8 to 1.0 × 10−8 M when measured by surface plasmon resonance. ScFv antibody fragment LA13-IIE3 showed best affinity, high long-term stability and remarkable resistance to denaturation. This scFv antibody fragment was coupled to a QCM sensor. CRP quantification in up to 15 samples sequentially measured on the same sensor with intermitting regeneration by buffer was demonstrated.
Keywords: C-reactive protein (CRP), single chain Fv (scFv), phage display, Quartz crystal microbalance (QCM)
Introduction
C-reactive protein (CRP) belongs to a group of serum proteins that are strongly upregulated during the acute phase of bacterial infections. Today, the use of fast and reliable diagnostics of CRP within the scope of inflammation, infectious diseases and tissue injury is without controversy.1 Moreover, elevated CRP levels of more than 1–3 mg/L, also termed high sensitivity CRP (or hs-CRP), has become the focus of attention because it is an independent predictor of future risk for cardiovascular events among healthy individuals.2,3 CRP is involved in host defense, scavenging and metabolic functions through its capacity for calcium-dependent binding to exogenous and autologous molecules containing phosphocholine (PC) and then activating the classical complement pathway by binding to C1q.4 CRP has at least two conformationally distinct isoforms, a native pentameric and a monomeric form. The latter is sometimes referred to as neoCRP. Monomeric CRP (mCRP), but not native pentameric CRP (also referred to as pCRP or nCRP), binds factor H, thereby directing this essential complement inhibitor to the surface of apoptotic and necrotic endothelial cells. It thus contributes to the safe removal of opsonized damaged cells and particles. Factor H–mCRP complexes enhance C3b inactivation both in the fluid phase and on the surface of damaged cells, and inhibit the production of pro-inflammatory cytokines, which counterbalances the function of the pentameric CRP.5 Calcium-dependent binding of pCRP to lipid membranes, including liposomes and cell membranes leads to a rapid partial structural change, forming a membrane bound pentameric intermediate, which significantly enhances complement fixation. This intermediate can detach from membranes to form monomeric CRP in solution, which exerts potent stimulatory effects on endothelial cells.6 A variety of CRP detection immunoassays have been developed to surrogate conventional erythrocyte sedimentation rate,7,8 but there is still need for a simple, fast and reliable bedside (point of care) test to determine a patient's inflammation status quickly and to provide quantitative data of the CRP serum level. These requirements have not been met by either microtiter-based immunoassays, which can take hours, or by semiquantitative lateral flow strip assays.9 Quartz crystal microbalance (QCM) sensors miniaturized to fit into microfluidic lab-on-chip systems10 coupled with CRP-specific antibodies may provide a suitable alternative.
In this study, we generated CRP-specific single chain (sc) Fv antibodies, optimized immobilization strategies for their coupling onto the sensor chip and demonstrated their use for repeated measurement cycles.
Results
Generation of CRP specific recombinant scFv antibody fragments
Prior to coupling to the panning support, the CRP antigen was analyzed by size exclusion chromatography, revealing a single peak at ~115 kDa (data not shown). As the calculated molecular weight of a CRP monomer is ~23 kDa, it can be assumed that the CRP was present in its homopentameric form. Recombinant scFv antibody fragments were isolated by phage display with three panning rounds on CRP immobilized on microtiter plates. The antibody phage library was generated using Hyperphage11 to provide oligovalent display in the first panning round, whereas the two subsequent rounds were done using M13K07 packaged phagemids. Identification of binders was achieved by using ELISA of E. coli culture supernatants containing soluble scFv fragments on immobilized CRP. DNA sequencing of eight ELISA positive phagemid clones revealed three unique scFv antibody clones. These three scFv clones LA13-IIE3, LA13-IIC3 and LA13-IID4 showed good binding to CRP at low antibody concentrations and no binding to BSA. For determination of their affinities, the scFv antibodies were produced in E. coli and purified by IMAC before being applied to a Biacore 2000 SPR system (Fig. 1A-C). Dissociation constants KD ranged from 2.7 × 10−8 M to 1.0 × 10−8 M with the scFv LA13-IIE3 showing the highest affinity (Fig. 1D). Moreover, the scFv LA13-IIE3 exclusively formed monomers as shown by size exclusion chromatography, which excludes multivalent binding to CRP (Fig. 2).

Figure 1. Affinity determination of the CRP specific scFv antibodies by SPR analysis. CRP was covalently coupled to a CM5 sensor chip. Sensorgrams of the CRP specific scFv antibody clones LA13-IIE3 (A), LA13-IIC3 (B) and LA13-IID4 (C) are shown. Four to five scFv concentrations were used to determine the affinity constants KD and off rates koff (D).
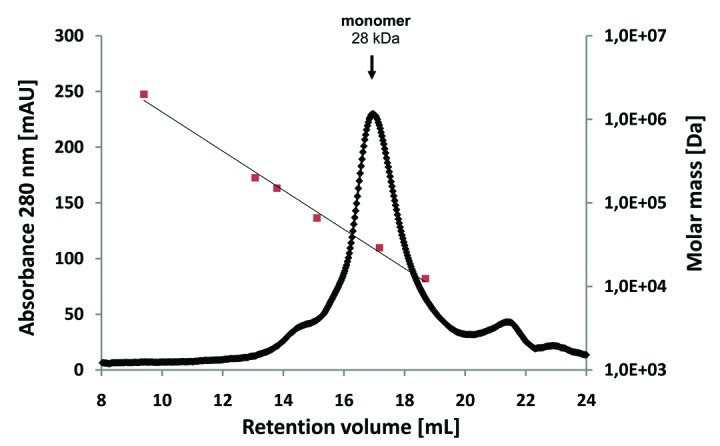
Figure 2. Size-exclusion chromatography of affinity-purified scFv LA13-IIE3 on a calibrated Superdex 200 10/300 GL column. Molar mass calibration (closed orange squares) was done with blue dextran (2,000 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), carbonic anhydrase (29 kDa) and cytochrome C (12.4 kDa).
Epitope binning
Epitope binning studies were performed to identify anti-CRP scFv antibody pairs that recognize different epitopes and do not interfere with each other during antigen binding (“sandwich pairs”). SPR assays were repeated with sequential injection of all possible combinations of two different scFv antibodies in the same run (Fig. 3). In this kind of assay, the SPR response signal will increase after injection of the second scFv antibody due to binding of both scFv to the sensor chip only if the two scFv antibodies recognize different epitopes without inhibiting each other. In contrast, if both scFv recognize the same epitope or interfere with each other during antigen binding, the signal will remain constant. Measurements were repeated by injecting scFv pairs in opposite order to assess the affect of steric hindrance of the scFv antibody that was bound first to CRP.

Figure 3. Epitope binning analysis of the CRP-specific scFv antibodies using SPR analysis. Overlay plots of SPR sensorgrams are arranged according to the scFv antibody injected first: LA13-IIE3 (A), LA13-IIC3 (B), and LA13-IID4 (C). All scFv antibodies were injected with saturating concentrations according to the CRP coupled CM5 sensor chip. Flow rate were kept constant throughout the measurement. The second injection of a different scFv antibody than the first one (run 4, 5, 9, 10, 14, and 15; red or blue solid lines) resulted only in an increased response signal if antigen binding of first and second scFv antibody did not interfere with each other. Injection of saturating concentrations of solely the first (runs 2, 7, and 12; gray dotted lines) or solely the second (runs 1, 6, and 11; black dashed lines) scFv antibody resulted into a response curve with typical dissociation and association pattern. Continuous injection of the first scFv antibody (runs 3, 8 and 13; green solid lines) were included as additional controls.
Sequential injection of LA13-IIC3 and LA13-IID4 did not result in a secondary mass increase on the Biacore chip independent of the order in which both scFv antibodies were injected. Therefore, LA13-IIC3 and LA13-IID4 recognize the same or an overlapping epitope (Fig. 3B and C). In contrast, injection of LA13-IIE3 as first scFv antibody followed by either LA13-IIC3 or LA13-IID4 led to a mass increase, indicating that LA13-IIE3 bound to another CRP epitope. Interestingly, injection in reverse order did not lead to any secondary mass increase if LA13-IIE3 was injected as second antibody fragment indicating some residual steric hindrance when LA13-IID4 or LA13-IIC3 are bound first to CRP (Fig. 3B and C; run 9 and 15). As a control, PBS was injected instead of first or second antibody and showed a typical dissociation pattern, whereas continuous injection of the first scFv antibody resulted in a constant response signal at the CRP coupled sensor.
Identification of the epitope structure
To further characterize the structure of the epitopes, scFv antibodies LA13-IIE3, LA13-IIC3, and LA13-IID4 were analyzed for their binding to linear epitopes. CRP was boiled in reducing SDS sample buffer, separated by SDS PAGE, and blotted onto PVDF membrane. Subsequent immunostaining with the scFvs revealed that all of the scFv antibodies specifically bound to a band in accordance to the calculated molar mass of monomeric CRP of ~23 kDa (Fig. 4). All three scFv antibodies can therefore be considered to recognize denatured CRP, which suggests that they bind to sequential epitopes.
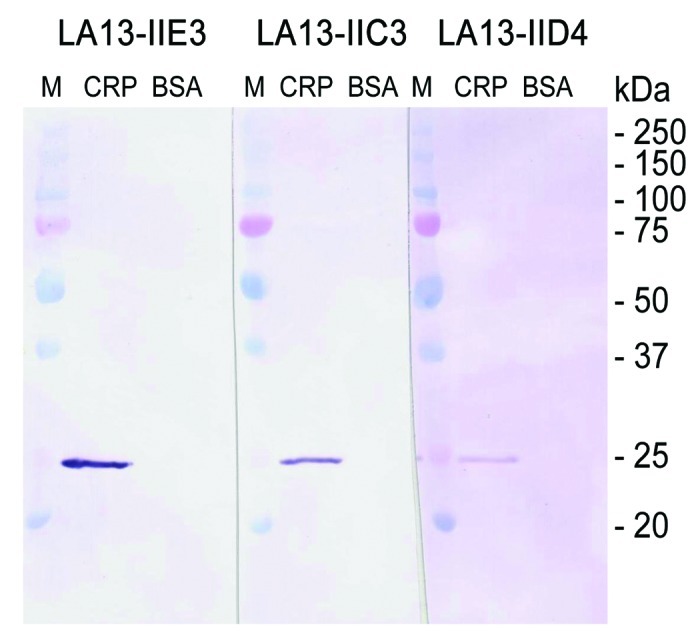
Figure 4. Immunoblot of CRP using scFv antibodies LA13-IIE3, LA13-IIC3 and LA13-IID4. CRP was prepared in SDS sample buffer (5 min, 98°C). A total of 150 ng CRP per lane was electrophoretically separated in a 12% (w/v) SDS polyacrylamide gel and transferred onto PVDF membrane. Immunostaining was performed with the CRP specific scFv antibodies LA13-IIE3, LA13-IID4, and LA13-IIC3 followed by the myc-tag specific mAb Myc1-9E10 and an AP conjugated secondary antibody conjugate. BSA was used as negative control.
Since LA13-IIE3, LA13-IIC3, and LA13-IID4 recognized sequential epitopes, these scFv antibodies could be subjected to detailed epitope mapping using an immobilized peptide spot arrays. Overlapping 15mer oligopeptides with 3 amino acid offset covering the entire human CRP protein sequence were synthesized on modified cellulose paper sheets in parallel spots12 and immunostained using the phage display-derived scFv fragments. Two independent experiments revealed that LA13-IIE3 stained two peptide spots both containing the core sequence NMWDFVLSPDEI (Fig. 5C). In contrast, LA13-IIC3 and LA13-IID4 recognized another epitope represented by the amino acid sequence IILGQEQDSFGG (Fig. 5A and B). These epitopes were further fine-mapped by alanine walking, a peptide spot analysis where variants of the epitopes with a single alanine substitutions per peptide for every side chain were analyzed (data not shown). The following amino acids side chains were identified as essential for the binding (underlined): NMWDFVLSPDEI (for LA13-IIE3), IILGQEQDSFGG (for LA13-IIC3) and IILGQEQDSFGG (for LA13-IID4). According to published crystal structures,4 they consist of α-helical and β-sheet secondary structure elements and are both at least partially surface accessible (Fig. 5D). Moreover, these epitopes are localized some distance from each other, which is in accordance with the observation that binding of scFv LA13-IIE3 to CRP was not inhibited by LA13-IIC3 or LA13-IID4, but only if LA13-IIE3 bound first to CRP as shown by SPR based epitope binning. Against that, in reverse order LA13-IIC3 or LA13-IID4 interfered with LA13-IIE3 antigen binding. Presumably, LA13-IIC3 or LA13-IID4 block the accessibility of the LA13-IIE3 epitope, which is located more inside the hole in the center of the CRP pentamer due to steric hindrance. Therefore, sandwich immunoassays using LA13-IIE3 as first antibody in combination with one of the other two scFvs are possible, but not if used in reverse order.

Figure 5. Epitope mapping of CRP specific scFv LA13-IIC3, LA13-IID4, LA13-IIE3. A series of overlapping 15mer oligopeptides covering the whole CRP protein sequence with an offset of three amino acids was synthesized onto filter membranes. These filter membranes were subsequently immunostained with CRP specific scFv antibodies LA13-IIC3 (A), LA13-IID4 (B), and LA13-IIE3 (C). Bound scFv antibodies were detected with α-myc-tag antibody and secondary antibody AP conjugate. Both epitopes are highlighted (yellow, space fill) in one subunit of the CRP pentamer structure (D). Epitope location based on structure (PDB ID: 1B09)4: peptide backbone (gray ribbon), side chains (lines), and polyhendron surface (points) are shown (visualization with 3D-Mol Viewer), epitopes indicated in yellow. Important amino acid residues of the epitopes identified by an additional alanine walk (data not shown) are underlined (* only IIC3)
Stability of scFv LA13-IIE3 and LA13-IID4
Stability of scFv fragments was tested using prolonged incubation at 37°C or repeated cycles of treatment with highly chaotropic GuHCl (Fig. 6). In the first experiment, two other scFvs were used as controls: the scFv IIB6 specific for human MUC1 was described to have a low stability of less the 24 h at 37°C;13 the second control scFv was derived from the hen egg white lysozyme specific antibody D1.3 and was chosen for its reportedly good stability.14 The scFv LA13-IIE3 showed extraordinary good stability after prolonged incubation at 37°C and also after treatment with GuHCl. It still retained 95% of its initial CRP binding activity after 15 d incubation at 37°C. Its antigen binding activity dropped below 50% only after more than 20 d incubation at 37°C (Fig. 6A). In contrast, the scFvs LA13-IIC3 and LA13-IID4 lost 40% and 70% of their initial antigen binding activity after 24 h incubation at 37°C (data not shown). The scFvD1.3 showed a drop of antigen binding after 10 d. None of the scFvs showed any unspecific binding to BSA in these experiments. After three sequential incubations with 3 M GuHCl, scFv LA13-IIE3 still retained 72% of its antigen binding activity. ScFv LA13-IID4 retained 65% of its initial binding activity. After 4x denaturation, their antigen binding activity further decreased to 50% and 25%, respectively (Fig. 6B).
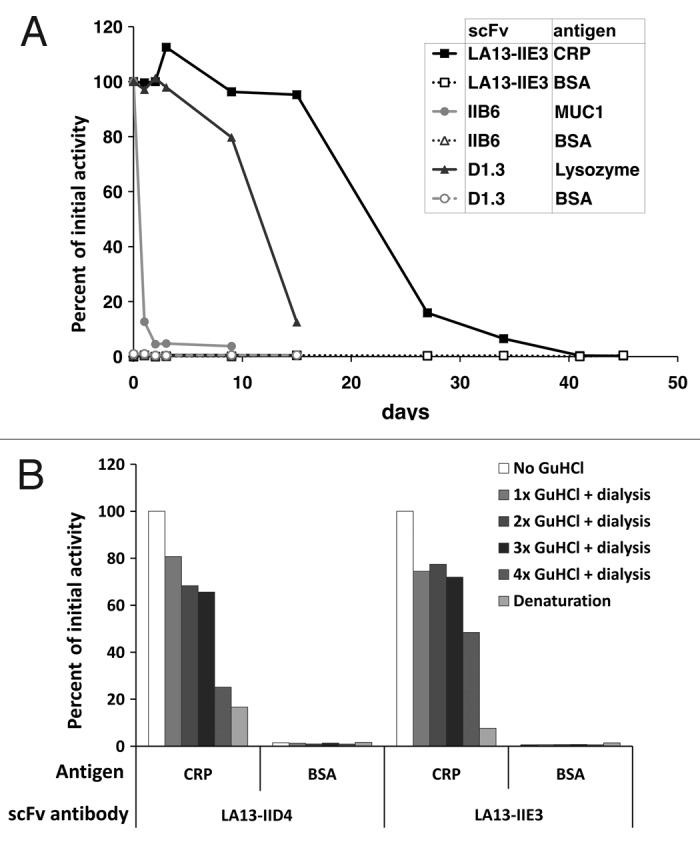
Figure 6. Stability tests of CRP specific scFvs. (A) Long-term stability of the scFv LA13-IIE3 was tested by incubation in PBS for up to 45 d at 37°C. Samples were analyzed by ELISA using CRP as antigen and BSA as control antigen. Freshly thawed scFv samples were used as references corresponding to 100% binding activity. For comparison two scFvs, the lysozyme specific scFv D1.3 and the mucin 1 specific scFv IIB6 were analyzed and tested to their antigens. Bound scFv antibody was detected with mouse α-myc-tag mAb and secondary antibody HRP conjugate. (B) Samples of the CRP specific scFvs LA13-IIE3 and LA13-IID4 were incubated with a final concentration of 3 M GuHCl for 30 min at room temperature followed by dialysis against PBS. The procedure was repeated several times and antigen binding activity was compared with untreated scFv as reference value.
CRP assays using scFv antibody coated QCM sensors
Before being applied to the gold surface of the QCM chip, scFv antibody coating and blocking conditions were optimized on gold sputtered microtiter plates allowing parallel analysis of a larger number of parameters (Fig. 7). SAM using cystamine activated by a glutaraldehyde crosslinker showed superior properties regarding the functional immobilization of scFv LA13-IIE3 compared with other SAMs using 11-MUA or DSP (Fig. 7). Although 11-MUA was able to immobilize a higher amount of scFv LA13-IIE3 protein, the CRP binding was almost completely lost in this procedure (Fig. 7D). Blocking with caseinate yielded the highest specific CRP signals compared with dry milk powder or BSA (Fig. 7A and C). To assess improved coupling to gold, a variant of the scFv fragment LA13-IIE3 containing a free cysteine at the carboxyterminal tag was constructed by subcloning into E. coli vector pOPE51.15 This scFv LA13-IIE3 variant showed lower recombinant production yield in E. coli and the additional unpaired cysteine did not lead to improved coupling with any of the three immobilization methods when tested on gold coated microtiter plates (Fig. 7B and C).

Figure 7. Optimizing conditions for scFv coupling to gold sputtered surface. Conditions for functional coupling of the anti-CRP capture scFv LA13-IIE3 to a gold-sputtered surface of a polypropylene microtiter plate (Gold-PP) were tested by sandwich ELISA using the antigen CRP and for detection anti-CRP mAb followed by a goat-anti mouse-HRP conjugate. Coupling was tested with SAMs using 11-MUA, DSP, or cystamine / glutaraldehyde (Cys/GA), or by direct incubation in combination with (A) different blocking conditions using milk powder (MP), BSA or caseinate or (B, C) by comparing unmodified scFv with the scFv-Cys variant containing a free C-terminal cysteine using (B) caseinate blocking and different SAMs, or (C) Cys/GA coupling and different blocking conditions. (D) Total amount of coupled scFv was detected by myc-tag specific mAb Myc1-9E10 was compared with the amount of functional scFv detected by CRP sandwich ELISA for different SAM coupling (casinate blocking). Polypropylene (PP) microtiter plates were used as control.
Based on these results, QCM sensors were prepared by coupling CRP specific scFv LA13-IIE3 antibody via cystamine/glutaraldehyde crosslinker SAM, and blocking was done with caseinate. The coupling and blocking steps were followed online by observing the corresponding frequency shifts. Both steps resulted in the expected frequency decrease indicating mass deposition on the chip surface (Fig. 8). After reaching constant signal baselines by rinsing the sensor with PBS, up to 32 injections of either CRP, BSA (negative control) or buffer were sequentially applied (Fig. 8). The observed frequency drop showed a good correlation to the concentration of the CRP in the sample at concentrations between 250 µg/mL and 1 mg/mL (Fig. 9A), while application of BSA did not result in a frequency drop, indicating binding. A run with 13 consecutive injections of 0.5 mg/mL CRP revealed a standard deviation of less than 7% between the sequential measurements (Fig. 9B).
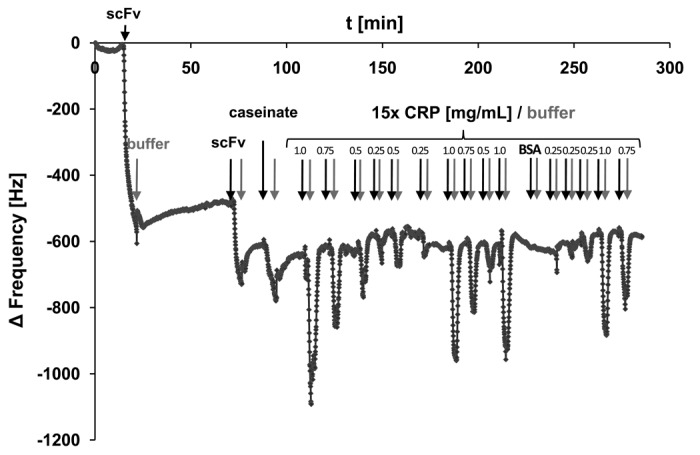
Figure 8. Preparation of the scFv LA13-IIE3 QCM microchip immunosensor and repeated CRP measurements. All injections of scFv, caseinate and CRP are indicated with black arrows whereas those of buffer (PBS) are indicated with gray arrows. The gold sputtered surface of the QCM sensor was activated with a cystamine / glutaraldehyde SAM followed by two injections of 20 and 60 µg/mL scFv LA13-IIE3, respectively. After washing with PBS 10 mg/mL caseinate was injected to block unspecific binding sites. After washing with PBS several measurement cycles were performed comprising injection of 0.25 to 1.0 mg/mL CRP followed by washing with PBS until baseline was retained. A total of 1.0 mg/mL BSA was measured to test unspecific frequency shifts, followed by new CRP measurements. Flow rate was kept at 34 µL/min. Sensor frequency was measured every 83 ms. A total of 20,766,111 Hz was subtracted to set the frequency before scFv coupling to 0 Hz.
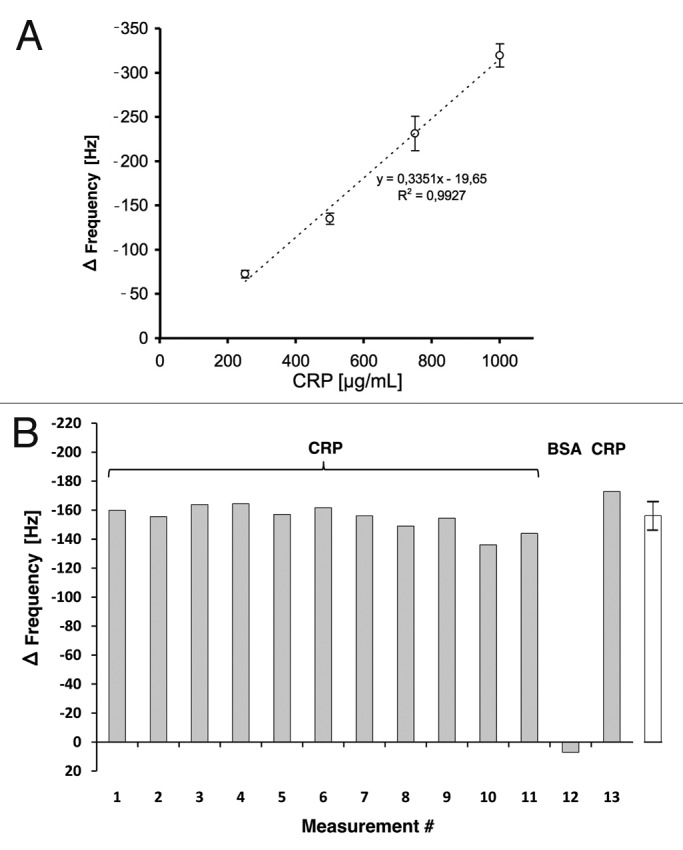
Figure 9. (A) Calibration of the scFv LA13-IIE3 QCM sensor. Measurements disturbed by air bubble peaks were excluded from the calculation of the linear regression equation (dotted line). Correlation coefficient R2 is indicated. Error bars represent standard deviations from triplicate measurements. (B) Repeated measurements to validate reproducibility of CRP specific frequency drops measured with the scFv LA13-IIE3 QCM immunosensor. A total of 12 injections of 0.5 mg/mL CRP and one injection of BSA (#12) used as negative control were applied to the same sensor chip using constant flow rate of 34 µL/min. Sensor frequency was measured every 83 ms. The maximum frequency change before and during CRP or BSA injection is shown. The right (white) bar represents the average frequency shift and standard deviation of all CRP measurements (#1–11 and #13).
Discussion
A QCM measurement does not require any additional label for detection because mass accumulation is directly detectable by a frequency shift.16 The use of QCM chips in combination with antibodies as highly specific antigen sensors would allow a label free (homogenous) measurement cycle within less than 30 min. Their properties would allow the implementation into point of care “lab-on-chip” devices suitable for online monitoring. In contrast to the current state of the art immunoassays like ELISA, this would vastly decrease the span between bedside blood collection, diagnostic assessment and start of treatment. Further, the repeated use of the same QCM immunosensor for several measurements may reduce costs compared with disposable systems, but, most importantly, would allow a calibration with predefined standards before or after the measurement of the patient sample. This, in contrast to other quick assays like lateral flow strips, promises to allow a truly quantitative determination. The use of the same QCM chip for sequential measurements of different samples, however, requires antibodies of exceptionally good stability.
In this study, we isolated recombinant scFvs antibody fragments with high affinity to two different epitopes on human CRP. The results of the epitope analysis suggest a spatial arrangement of the binding sites that is conceivable from the known three dimensional structure of CRP. In detail, the epitopes do not seem to be masked in the assembled CRP pentamer and located in sufficient distance to each other to explain the sandwich pair analysis results. An interesting observation is that while scFv LA13-IIE3 binding to CRP was not inhibited by LA13-IIC3 or LA13-IID4, when applied in reverse order, LA13-IIC3 or LA13-IID4 interfered with LA13-IIE3 antigen binding. As the pentamer forms a sort of ring structure, LA13-IIC3 or LA13-IID4 may block the access of the LA13-IIE3 epitope to the “hole” in the center of the CRP pentamer before it can assume its final binding position. A structural change of CRP caused after LA13-IIC3 or LA13-IID4 binding to CRP is also not excluded.
The very good stability of scFv LA13-IIE3 in solution has been observed only for a small number of other scFv antibodies so far. This stability promises to allow long shelf lives, another essential prerequisite for a successful use in a robust point of care diagnostic system. Even after three consecutive, rude treatments of the scFvs LA13-IIE3 and LA13-IID4 with chaotropic agent, the antigen binding activity was reduced by less than 35%. In the future, more stringent washing steps or even regeneration cycles using a variety of reagents that usually denature antibodies may be tested, and, if successful, these would allow the use of the same sensor chip for more than the 16 consecutive measurement cycles already demonstrated in this study. An interesting observation is that the CRP dissociated from the QCM chip surface coated with scFv LA13-IIE3 faster than in the measurement done by plasmon resonance. To explain this, it may be speculated that either CRP or the scFv antibody undergo structural changes upon coupling to the chip, or that the gold surface of the sensor influenced the surface deposition in a different way compared with the dextran matrix used in surface plasmon resonance assays. The high frequent vibrations of the QCM sensor may also influence the antibody CRP interaction, e.g., by increasing the off rate. Although full understanding of the reason is lacking, the observed frequency shift upon binding correlated very well with the amount of CRP applied, so the rapid dissociation facilitated the sequential measurement of up to 16 different samples (including one BSA control measurement) in a short time span. Whether this is a feature specific for the CRP antigen or this antibody, or a more general effect provided by these antibodies, the scFv format, the sensor surface chemistry or its layout remains to be tested in the future.
Clinically-relevant plasma concentrations of CRP indicating pathologic processes are about 10–40 mg/L in mild inflammation and viral infections, about 40–200 mg/L in active inflammation or bacterial infection, and can rise up to 500 mg/L in severe cases, whereas in healthy people, plasma concentrations of CRP are normally below 1–5 mg/L.17,18 With a current sensitivity only covering the upper part of this range, our QCM-antibody sensor system needs to be improved. Moreover, direct use of serum samples on this QCM immunosensor caused unstable and high background frequencies masking CRP specific signals lower than 1 g/L (data not shown). These issues could be addressed by a variety of approaches, e.g., implementation of an affinity chromatography cell for concentration and purification of CRP from the serum upstream from the QCM immunosensor in the same microfluidic lab on chip system.19, 20 This would not only alleviate the impact of matrix effects of serum samples on the accuracy of the measurements, but theoretically allows use of the system even for the detection of elevated CRP levels at low concentrations described as a predictive value for cardiovascular diseases.2,3 Sensitivity could also be enhanced by further engineering of the detector antibody to increase its affinity. Here, a variety of methods have been successfully applied and resulted in affinity increases of several orders of magnitude,21,22 with some examples significantly exceeding the best affinities achieved from conventional monoclonal antibodies.
The recombinant single chain antibody fragment scFv LA13-IIE3 generated by antibody phage display showed high long-term stability and robustness against chaotropic conditions. This scFv antibody fragment was successfully used in QCM immunosensors for up to 15 repeated CRP quantifications using the same chip. In the future, an improved version of the combination of recombinant antibodies with a miniaturized flow-through QCM sensor may be developed into a lab on chip system for rapid high accuracy point of care determination of CRP levels. 20
Materials and Methods
Selection of scFvs against human CRP by phage display
CRP specific scFv antibody fragments were isolated from a scFv antibody fragment library23 by panning in polystyrole microtiter stripes as described.24 Briefly, Maxisorp microtiter stripe wells (Nunc) were coated with 100 µL per well 1 µg/mL human recombinant CRP (BiosPacific) in phosphate buffered saline (PBS: pH 7.4, 10x stock solution contains 1.37 M NaCl, 26 mM KCl, 80 mM Na2HPO4, 15 mM KH2PO4) overnight at 4°C. Antigen coated wells were blocked with MPBST consisting of 2% (w/v) skim milk powder (Roth) in PBST [PBS supplemented with 0.05% (v/v) Tween-20 (Serva)] for 1 h at room temperature (RT), followed by three washing steps with PBST. A total of 1011 to 1012 cfu (colony forming units) phage of the antibody gene libraries were pre-incubated in blocked wells with MPBST for 1 h at RT to pre-adsorb unspecific binding phage. Supernatant was transferred into antigen-coated wells and incubated for 2 h at RT. Non-bound antibody phage were removed by ten stringent washing steps using the ELISA washer Columbus Pro (TECAN). In following panning rounds the number of washing steps was increased (second round: 20 washing steps, third panning round: 30 washing steps). Finally, bound phage were eluted by incubation with 200 µL/well of 10 µg/mL trypsin solution for 30 min at 37°C. A total of 10 µL of eluted phage was used for phage titration, whereas the residual eluate was used for re-infection into E. coli XL1-Blue-MRF’ and phage production as described.25 Briefly, XL1-Blue MRF’ were cultured overnight in 2xYT medium [16 g/L tryptone, 10 g/L yeast extract (Difco, Voigt), 5 g/L NaCl) supplemented with 50 µg/mL tetracycline (2xYT-T) at 250 rounds per minute (rpm) and 37°C (Multitron incubation shaker, Infors, CO2)]. Next day, E. coli cells were diluted 1/100 and grown in 50 mL fresh 2xYT medium until an optical density at 600 nm (OD600) of 0.5 at 37°C and 250 rpm. These exponentially growing E. coli were then infected with eluted phage by incubation at 37°C for 2x 30 min without and with agitation, respectively. Infected bacteria were plated onto YT-GA agar plates [2xYT medium supplemented with 100 mM glucose (G), 100 µg/ml ampicillin (A) and 15 g/L agar] and incubated overnight at 37°C. Colonies were harvested by resuspension in 5 mL 2xYT-GA medium using a Drigalsky spatula. A total of 200 µL of this bacterial suspension was inoculated in 50 mL 2xYT-GA at 37°C at 250 rpm. After reaching exponential growth phase (OD600 ~0.5) 5 mL of this bacterial suspension were infected with 5 × 1010 cfu of M13K07 helper phage. Following, infected bacterial cells were harvested by centrifugation and resuspended in 30 mL 2xYT-AK medium (2xYT with 100 µg/mL ampicillin and 50 µg/mL kanamycin). Phage production was performed at 30°C and 250 rpm overnight. Bacterial cells were removed by centrifugation and phage were precipitated from supernatant by adding 1/5 volume of a 20% (w/v) polyethylene glycol (PEG) 6000 solution supplemented with 2.5 M NaCl and 1 h incubation in an overhead shaker at 4°C. Phage precipitate was obtained by centrifugation for 1 h at 3,200x g and 4°C. Phage were resuspended in 10 mM TRIS-HCl pH7.5, 20 mM NaCl and 2 mM EDTA (EDTA) and used for the next panning rounds.
ScFv production and purification
After screening and DNA sequencing, unique CRP specific scFv antibody gene fragments were subcloned by NotI and NcoI into the scFv expression vector pOPE10115 providing fusion with gene sequences encoding hexa-histidine and myc tag (mAb Myc1-9E10 epitope). For scFv production 300 mL 2xYT-GA medium was inoculated with an overnight culture of XL1-Blue MRF' carrying CRP-specific scFv antibody clones (initial OD600 < 0.1). Cells were grown at 37°C and 250 rpm until OD600 ~0.5. Expression was induced with 50 µM IPTG and scFv antibody production was performed for 12 h at 30°C and 250 rpm. Cells were spun down at 13,000x g for 20 min. ScFv proteins were precipitated from the supernatant by gradually dissolving 40 g ammonium sulfate per 100 mL culture volume and centrifugation for 20 min at 13,000x g. The precipitate was resuspended in PBS (1/30 of the culture volume). In addition, scFvs were also prepared from the bacterial periplasma by resuspending the cell pellet in 1/10 volume PE buffer (50 mM TRIS-HCl pH 8, 20% (w/v) sucrose, 1 mM EDTA) and vigorous shaking for 20 min at 4°C, followed by centrifugation for 10 min at 3,220x g. Both, supernatant and periplasmic fractions were used for purification. ScFv were purified by affinity chromatography using immobilized metal affinity chromatography (IMAC) using 0.5 mL Chelating Sepharose Fast Flow (GE Healthcare, Munich, Germany) referring to the manufacturer's instructions.
Sodium dodecyl sulfate polyacrylamide elecrophoresis (SDS-PAGE) and immunoblot
CRP-specific scFv antibodies were prepared under reducing conditions in Laemmli sample buffer for 5 min at 95°C, then separated by SDS-PAGE and blotted onto a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with MPBST for 1 h at RT. The myc-tag of the scFv was detected with the mouse α-myc-tag monoclonal antibody (mAb Myc1-9E10, 1 µg/mL) for 1.5 h at RT, followed by three washing steps with PBST. Goat α-mouse IgG (Fc-specific) antibody alkaline phosphatase (AP) conjugate (Sigma, Taufkirchen, Germany; 1:20,000) was used for detection and stained by nitro-blue tetrazolium chloride (NBT)/bromo-4-chloro-3-indolylphosphate toluidine (BCIP).
Stability test of scFv antibodies
Stability test of purified scFvs was performed by preparing identical aliquots (20–50 µg/mL scFv) which were stored at -80°C. Samples were thawed at different time points and incubated for up to 45 d at 37°C. Finally, all samples were tested by antigen ELISA (see below). The percentage of functional scFv was calculated by comparing the absorption measured in antigen ELISA in comparison to the freshly thawed scFv sample (100%).
Stability of scFv protein in the presence of chaotropic agents was determined by repetitive denaturation of scFv samples with guanidinium hydrochloride (GuHCl) in a final concentration of 3 M followed by dialysis to PBS overnight. Samples were denaturated up to four times. Antigen binding activity and specificity of the scFv antibodies were determined by antigen ELISA as described below in comparison to non-treated samples. Completely denatured scFv (in 3 M GuHCl without dialysis) was used to determine the background in the ELISA. Bovine serum albumin (BSA) was used as control antigen to exclude unspecific binding of denatured scFv antibody.
Enzyme linked immunosorbent assay (ELISA)
CRP specific antigen binding of soluble scFv fragments were tested for the screening as well as for the stability tests by antigen ELISA.26 Briefly, Maxisorp microtiter plates (Nunc) were coated with 100 µL 1 µg/mL human recombinant CRP in PBS, pH 7.4 per well overnight at 4°C. Coated wells were blocked with MPBST for 1 h at RT, followed by three washing steps with PBST. A total of 100 µL/well scFv containing supernatant from screening production or scFv samples from the stability test diluted 1:5 in MPBST were incubated for 1.5 h at RT, followed by three washing steps with PBST using an ELISA washer. Bound scFvs were detected with 100 µL/well mouse α-myc-tag mAb Myc1-9E10 (1:1000) and polyclonal goat α-mouse IgG (Fab-specific) antibody horse radish peroxidase (HRP) conjugate (1:20,000). Color reaction was performed using TMB (3,3′,5,5′-tetramethylbenzidine) substrate and stopped by adding 100 µL 0.5 M sulphuric acid. Absorbance at 450 nm (A450) was measured using the microtiter plate reader Sunrise (Tecan). The absorbance at 620 nm (A620) was subtracted.
Surface plasmon resonance (SPR) analysis
Affinities of CRP-specific scFv were determined using SPR in a Biacore 2000 (GE Healthcare).27 Recombinant CRP was immobilized by amine coupling to a CM5 sensor chip28 corresponding to a maximum of 200 response units (RU) according to the manufacturer's instructions. A flow rate of 30 µL/min was maintained during all measurements. For each scFv antibody clone five different concentration (1, 3, 10, 30 and 100 nmol in PBS) were tested. Association was measured for 100 sec and dissociation for up to 600 sec. After measurement of each scFv dilution the chip was regenerated with 50 mM NaOH and 10 µL/min flow rate for 30 sec. Dissociation constants (KD) were calculated by using the BIAevaluation software (Langmuir 1:1 fitting).
Analytical size exclusion chromatography
A total of 60 µg purified scFv LA13-IIE3 was injected onto a calibrated Superdex 10/300 GL column using the Äkta Purifier fast performance liquid chromatography system (GE Healthcare). The column was equilibrated in PBS, pH 7.4, and the flow-rate was kept constant at 0.5 mL/min.
Epitope binning
To determine if the isolated scFv clones recognize different epitopes and if binding of different antibodies interferes with each other, epitope binning was performed by SPR analysis using the Biacore 2000 (GE Healthcare). CRP was coupled to CM5 sensor chips as described above. A concentration of 2.5 µg/mL first scFv antibody was injected with a flow rate of 30 µL/min until saturation, followed by co-injection of the second scFv antibody in the same concentration and with the same flow rate. Measurements were performed using the feature “coinject” of the Biacore 2000. All scFv antibody clones were compared against each other and also by switching the order. Controls experiments were run by injecting running buffer instead of the first or the second scFv, respectively.
Epitope mapping using a peptide spot array
Epitope mapping of the CRP-specific scFv antibodies was performed using an array of immobilized 15mer oligopeptides covering the whole human CRP by overlapping with an off-set of three amino acids (kindly provided by Ronald Frank, HZI Braunschweig, Germany). The CRP peptide membrane was washed three times with Tris buffered saline (TBS, 150 mM NaCl, 20 mM Tris, pH 7.4), blocked with 2% (w/v) skim milk powder in TBST [TBS, 0.05% (v/v) Tween-20] for 1 h at RT and incubating with each of the scFv antibodies for 1.5 h at RT on a rocket table. After three washing steps with TBST bound scFv antibodies were detected with 100 µL mouse α-myc-tag mAb Myc1-9E10 (1:500) and polyclonal goat α-mouse IgG (Fab-specific) alkaline phosphatase (AP) conjugate (1:10,000) washed twice with each TBS and citrate buffered saline (CBS, 138 mM NaCl, 2.67 mM KCl, 10 mM citric acid, pH 7.0) and visualized with staining solution [10 mL CBS, 50 µL 1 M MgCl2, 40 µL 6% (w/v) BCIP diluted in DMF, 60 µL 5% (w/v) 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid (MTT) diluted in 70% (v/v) DMF]. Reaction was stopped by washing twice with PBS. The membrane was stripped for re-use as described.12
Coupling of scFv to a gold surface
To identify the optimal parameters for coupling functional scFv antibodies to the gold surface of the QCM sensor, polypropylene (PP) microtiter plates (Greiner Bio-one) were gold-sputtered and either used directly (non-treated) or prepared self-assembling monolayers (SAM) using 5 mM 11-mercaptoundecanoic acid (11-MUA, in ethanol) followed by N-hydroxysuccinimide (NHS) / 1-ethyl-3-(3-dimethyl–aminopropyl) carbodiimide (EDC) activation, 20 mM dithiobis-succinimidylpropionate (DSP, in acetone), or 20 mM cystamine in combination with 2.5% (v/v) glutaraldehyde crosslinker. After three washing steps with H2O the scFv LA13-IIE3 was incubated for 1 h at RT. Then different blocking solutions [milk powder (MP), BSA or caseinate] were incubated for 1 h at RT. After three washing steps, functional scFv LA13-IIE3 was detected by capturing CRP (1 h, RT), followed by detection with the mouse α-CRP monoclonal antibody (mAb, clone 4C28, C6, Diasys, Holzheim) and a goat-anti-mouse-IgG HRP conjugate. Total scFv LA13-IIE3 was detected using the α-myc-tag mAb Myc1-9E10. Color reactions with TMB and measurements were performed as described above.
QCM sensor chip
The QCM sensor was constructed and embedded into a flow cell made of polydimethylsiloxane (PDMS) to optimize the flow conditions as described.10,29,30 The assembly of the quartz resonator and connection to the measuring system is also described there.
Coupling of the scFv antibody LA13-IIE3 on the QCM sensor chip
A total of 20–60 µg/mL of the CRP-specific scFv LA13-IIE3 was immobilized via a cystamine/glutaraldehyde SAM on the gold electrode of the quartz crystal. Briefly, the quartz sensor was washed three times with H2O and incubated with 20 mM cystamine overnight. After three washing steps with H2O the sensor was incubated with 2.5% (v/v) glutaraldehyde for 2 h.31 To immobilize the scFv to the activated SAM 100 µL of the scFv antibody fragment solution was pumped through the sensor cell with 34 µL/min followed by a PBS washing step. This procedure was monitored by measuring the frequency shift. All steps were done at RT.
CRP measurement using the antibody QCM sensor
Ten mg/mL caseinate was used for saturation of free binding sites after immobilization of the scFv LA13-IIE3 on QCM sensor chip. Measurements were performed using 100 µL of different concentrations of CRP in PBS (0.25–1.0 mg/mL) injected into the prepared QCM with a flow rate of 34 µL/min. PBS was used as running buffer. The measurements were performed at RT. As negative control, 1.0 mg/mL BSA solution was injected instead of the CRP solution.
Acknowledgments
We gratefully acknowledge the financial support by the German Research Foundation (DFG, SFB 578). We also thank Ronald Frank for providing the immobilized peptide spot membranes for epitope mapping. One of the authors (S.B.) gratefully acknowledges the financial support of the Volkswagen Foundation.
Disclosure of Potential Conflicts of Interest
This project was supported by the SFB578 of the Deutsche Forschungsgemeinschaft (Subproject D2). S.B. was supported by Volkswagenstiftung. Financial funding had no influence of the design of experiments and the interpretation of data in this study.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/22374
References
- 1.Kurosawa S, Nakamura M, Park J-W, Aizawa H, Yamada K, Hirata M. Evaluation of a high-affinity QCM immunosensor using antibody fragmentation and 2-methacryloyloxyethyl phosphorylcholine (MPC) polymer. Biosens Bioelectron. 2004;20:1134–9. doi: 10.1016/j.bios.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 2.Clearfield MB. C-reactive protein: a new risk assessment tool for cardiovascular disease. J Am Osteopath Assoc. 2005;105:409–16. [PubMed] [Google Scholar]
- 3.Verma S, Yeh ET. C-reactive protein and atherothrombosis--beyond a biomarker: an actual partaker of lesion formation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1253–6, discussion R1257-8. doi: 10.1152/ajpregu.00170.2003. [DOI] [PubMed] [Google Scholar]
- 4.Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7:169–77. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 5.Mihlan M, Hebecker M, Dahse H-M, Hälbich S, Huber-Lang M, Dahse R, et al. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol Immunol. 2009;46:335–44. doi: 10.1016/j.molimm.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Ji S-R, Wu Y, Zhu L, Potempa LA, Sheng F-L, Lu W, et al. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m) FASEB J. 2007;21:284–94. doi: 10.1096/fj.06-6722com. [DOI] [PubMed] [Google Scholar]
- 7.Simon L, Gauvin F, Amre DK, Saint-Louis P, Lacroix J. Serum procalcitonin and C-reactive protein levels as markers of bacterial infection: a systematic review and meta-analysis. Clin Infect Dis. 2004;39:206–17. doi: 10.1086/421997. [DOI] [PubMed] [Google Scholar]
- 8.Urbach J, Shapira I, Branski D, Berliner S. Acute phase response in the diagnosis of bacterial infections in children. Pediatr Infect Dis J. 2004;23:159–60. doi: 10.1097/01.inf.0000115735.78960.a4. [DOI] [PubMed] [Google Scholar]
- 9.Leung W, Chan CP, Rainer TH, Ip M, Cautherley GWH, Renneberg R. InfectCheck CRP barcode-style lateral flow assay for semi-quantitative detection of C-reactive protein in distinguishing between bacterial and viral infections. J Immunol Methods. 2008;336:30–6. doi: 10.1016/j.jim.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Michalzik M, Wilke R, Büttgenbach S. Miniaturized QCM-based flow system for immunosensor application in liquid. Sens Actuators B Chem. 2005;111-112:410–5. doi: 10.1016/j.snb.2005.03.048. [DOI] [Google Scholar]
- 11.Rondot S, Koch J, Breitling F, Dübel S. A helper phage to improve single-chain antibody presentation in phage display. Nat Biotechnol. 2001;19:75–8. doi: 10.1038/83567. [DOI] [PubMed] [Google Scholar]
- 12.Frank R, Dübel S. Analysis of protein interactions with immobilized Peptide arrays synthesized on membrane supports. CSH Protoc. 2006;2006:2006. doi: 10.1101/pdb.prot4566. [DOI] [PubMed] [Google Scholar]
- 13.Thie H, Toleikis L, Li J, von Wasielewski R, Bastert G, Schirrmann T, et al. Rise and fall of an anti-MUC1 specific antibody. PLoS One. 2011;6:e15921. doi: 10.1371/journal.pone.0015921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villani ME, Morea V, Consalvi V, Chiaraluce R, Desiderio A, Benvenuto E, et al. Humanization of a highly stable single-chain antibody by structure-based antigen-binding site grafting. Mol Immunol. 2008;45:2474–85. doi: 10.1016/j.molimm.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Schmiedl A, Breitling F, Winter CH, Queitsch I, Dübel S. Effects of unpaired cysteines on yield, solubility and activity of different recombinant antibody constructs expressed in E. coli. J Immunol Methods. 2000;242:101–14. doi: 10.1016/S0022-1759(00)00243-X. [DOI] [PubMed] [Google Scholar]
- 16.Sauerbrey G. Verwendung von Schwingquarzen zur Wagung dunner Schichten und zur Mikrowagung. Zeitschrift für Physik. 1959;155:206–22. doi: 10.1007/BF01337937. [DOI] [Google Scholar]
- 17.Deodhar SD. C-reactive protein: Clinical applications in monitoring disease activity. Clin Immunol Newsl. 1991;11:138–43. doi: 10.1016/0197-1859(91)90036-R. [DOI] [Google Scholar]
- 18.Clyne B, Olshaker JS. The C-reactive protein. J Emerg Med. 1999;17:1019–25. doi: 10.1016/S0736-4679(99)00135-3. [DOI] [PubMed] [Google Scholar]
- 19.Michalzik M, Balck A, Büttgenbach S, Al-Halabi L, Hust M, Dübel S. Proc of the XX Eurosensors Conference. Proc. of the XX Eurosensors Conference. Göteborg, Sweden, 2006:420–421. [Google Scholar]
- 20.Balck A, Michalzik M, Al-Halabi L, Dübel S, Büttgenbach S. Design and fabrication of a lab-on-a-chip for point-of-care diagnostics. 2011;127:102–111. [Google Scholar]
- 21.Thie H, Voedisch B, Dübel S, Hust M, Schirrmann T. Affinity maturation by phage display. Methods Mol Biol. 2009;525:309–22, xv. doi: 10.1007/978-1-59745-554-1_16. [DOI] [PubMed] [Google Scholar]
- 22.Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–54. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Wildt RM, Mundy CR, Gorick BD, Tomlinson IM. Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat Biotechnol. 2000;18:989–94. doi: 10.1038/79494. [DOI] [PubMed] [Google Scholar]
- 24.Hust M, Toleikis L, Dübel S. Handbook of therapeutic antibodies. Weinheim: Wiley-VCH, 2007:45–68. [Google Scholar]
- 25.Hust M, Dübel S, Schirrmann T. Selection of recombinant antibodies from antibody gene libraries. Methods Mol Biol. 2007;408:243–55. doi: 10.1007/978-1-59745-547-3_14. [DOI] [PubMed] [Google Scholar]
- 26.Schütte M, Thullier P, Pelat T, Wezler X, Rosenstock P, Hinz D, et al. Identification of a putative Crf splice variant and generation of recombinant antibodies for the specific detection of Aspergillus fumigatus. PLoS One. 2009;4:e6625. doi: 10.1371/journal.pone.0006625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schier R, McCall A, Adams GP, Marshall KW, Merritt H, Yim M, et al. Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site. J Mol Biol. 1996;263:551–67. doi: 10.1006/jmbi.1996.0598. [DOI] [PubMed] [Google Scholar]
- 28.Jönsson U, Fägerstam L, Ivarsson B, Johnsson B, Karlsson R, Lundh K, et al. Real-time biospecific interaction analysis using surface plasmon resonance and a sensor chip technology. Biotechniques. 1991;11:620–7. [PubMed] [Google Scholar]
- 29.Michalzik M, Balck A, Büttgenbach S, Al-Halabi L, Hust M, Dübel S. Berlin, Offenbach: VDE Verlag, 2007:S. 939–942. [Google Scholar]
- 30.Michalzik M, Wendler J, Rabe J, Büttgenbach S, Bilitewski U. Development and application of a miniaturised quartz crystal microbalance (QCM) as immunosensor for bone morphogenetic protein-2. Sens Actuators B Chem. 2005;105:508–15. doi: 10.1016/j.snb.2004.07.012. [DOI] [Google Scholar]
- 31.Michalzik M, Al-Halabi L, Balck A, Dübel S, Büttgenbach S. Development of on Chip Devices for Life Science Applications. International Journal of Engineering. 2009;3:148–58. [Google Scholar]