

Abstract
Here, we describe a new class of multivalent and multispecific antibody-based reagents for therapy. The molecules, termed “trimerbodies,” use a modified version of the N-terminal trimerization region of human collagen XVIII noncollagenous 1 domain flanked by two flexible linkers as trimerizing scaffold. By fusing single-chain variable fragments (scFv) with the same or different specificity to both N- and C-terminus of the trimerizing scaffold domain, we produced monospecific or bispecific hexavalent molecules that were efficiently secreted as soluble proteins by transfected mammalian cells. A bispecific anti-laminin x anti-CD3 N-/C-trimerbody was found to be trimeric in solution, very efficient at recognizing purified plastic-immobilized laminin and CD3 expressed at the surface of T cells, and remarkably stable in human serum. The bispecificity was further demonstrated in T cell activation studies. In the presence of laminin-rich substrate, the bispecific anti-laminin x anti-CD3 N-/C-trimerbody stimulated a high percentage of human T cells to express surface activation markers. These results suggest that the trimerbody platform offers promising opportunities for the development of the next-generation therapeutic antibodies, i.e., multivalent and bispecific molecules with a format optimized for the desired pharmacokinetics and adapted to the pathological context.
Keywords: antibody engineering, multivalent antibodies, bispecific antibodies, collagen, trimerbody
Introduction
Monoclonal antibodies (mAbs) are one of the fastest growing classes of therapeutic agents. Currently, more than 30 mAbs have been approved by regulatory agencies for clinical use,1 but conventional unmodified mAbs have limitations, such as low tumor-to-blood ratio, due to long serum half-life and limited tissue penetration, and specificity for a single antigen epitope.2 The latter is a particularly important aspect because many diseases are multifactorial, involving multiple ligands, receptors and signaling cascades. Consequently, blockade of different pathological factors and pathways may result in improved therapeutic efficacy.3
To circumvent the limitations of current mAbs, substantial efforts have been devoted to the development of the next wave of antibody-based reagents for therapy, i.e., multivalent and multispecific molecules that block two or more relevant targets, with a format optimized for the desired pharmacokinetics and adapted to the pathological context.4 Conversion of monovalent antibody fragments (Fab, scFv, or single-domain antibody), into multivalent formats increases functional affinity, decreases dissociation rates when bound to cell-surface receptors or polyvalent antigens, and enhances biodistribution.5
Monovalent antibody fragments have been engineered into multimeric conjugates using either chemical or genetic cross-links. The most common strategy to create multimeric IgG-like formats has been the engineering of fusion proteins in which the antibody fragment makes a complex with homodimerization proteins (e.g., ZIP miniantibody,6 scFv-Fc antibody7 and minibody8). A different strategy to multimerize antibody fragments is based on the reduction of the interdomain linker length (0–5 residues) to generate bivalent, trivalent or tetravalent antibodies (referred to as diabody, triabody or tetrabody, respectively).9 Strong protein-ligand interactions have been also used to make other multimeric non-IgG-like formats. For example, the ribonuclease barnase and its inhibitor barstar,10 TNFα11,12 streptavidin-biotin,13 and the dock-and-lock method (DNL) in which antibody fragments are fused to the regulatory subunit of the cAMP-dependent protein kinase A and the anchoring domain from A-kinase anchor protein.14
We recently described the in vitro and in vivo properties of a multivalent antibody made by fusing a trimerization (TIE) domain to the C-terminus of a scFv fragment. TIE domains are composed of the N-terminal trimerization region of collagen XVIII NC1 or collagen XV NC1 flanked by a flexible linker.15-17 The new antibody format, termed “trimerbody” [(scFv-NC1)3; 110 kDa] exhibited excellent antigen binding capacity and multivalency, which provided them with a significant increase in functional affinity and therefore enhanced binding capacity and slower dissociation rate.16,17
In this study, we used the trimerbody platform technology to create hexavalent molecules. By fusing scFv fragments to both N- and C-terminus of a TIEXVIII domain, monospecific or bispecific, hexavalent-binding trimerbodies were produced. Recombinant N/C-trimerbodies were efficiently secreted as soluble proteins by transfected human HEK-293 cells, and were able to recognize their cognate antigen with high affinity and specificity.
Results
Design, expression and functional characterization of scFv-based N-terminal, C-terminal and N/C-terminal trimerbodies
We have previously shown that fusion of a TIE domain to the C-terminus of a scFv fragment confers a trimeric state to the fused antibody.15-17 Each TIE domain is composed of the N-terminal trimerization region of collagen XVIII NC1 (TIEXVIII) or collagen XV NC1 (TIEXV) flanked by a flexible linker (Fig. 1). Purified N-terminal scFv-based trimerbodies (N-trimerbodyXVIII or N-trimerbodyXV) are trimeric in solution, and exhibit excellent antigen binding capacity.16,17

Figure 1. Schematic diagram showing the genetic constructs used in the production of trimerbody molecules. (A). All constructs bear a TIE domain composed of the N-terminal trimerization region of collagen XVIII NC1 (red box) flanked by one or two flexible linkers (yellow box). The trimerbody gene constructs contain a heterologous signal peptide from the oncostatin M (gray box), the scFv gene (VH and VL domains joined by a flexible linker) and a TIEXVIII domain. Arrows indicate the direction of transcription. His6myc tag (purple box) appended was for immunodetection. (B). Schematic representation of the scFv-based N-, C- and N/C-terminal trimerbodies.
In this study, we created a new generation of scFv-based trimerbodies by fusion of the LM111-specific L36 scFv gene to the C-terminus of a TIEXVIII domain (C-trimerbodyXVIII), or to both N- and C-terminus of a TIEXVIII domain (N/C-trimerbodyXVIII) (Fig. 1). Recombinant L36 scFv-based N-trimerbodyXVIII (L36N), C-trimerbodyXVIII (CL36) and N/C-trimerbodyXVIII (L36N/CL36) molecules were efficiently secreted as soluble proteins by transfected human HEK-293 cells, and were able to recognize their cognate antigen with high affinity and specificity (Fig. 2A). Western blot analysis demonstrated that under reducing conditions L36 scFv-based trimerbodies are single chain type molecules with masses of 36.5 kDa (L36N), 36.7 kDa (CL36) and 63.9 kDa (L36N/CL36). Remarkably, the culture medium containing the N/C-trimerbodyXVIII displayed a higher ELISA signal than the one with N-trimerbodyXVIII in spite of the higher yield observed in the culture medium of cells secreting the N-trimerbodyXVIII (L36N: 21 μg/ml × 105 cells/72 h vs. L36N/CL36: 8 μg/ml × 105 cells/72 h) (Fig. 2A and B).

Figure 2. Characterization of recombinant trimerbodies. The presence of secreted scFv-based trimerbodies in the conditioned media from gene-modified HEK-293 cells was demonstrated by western blot analysis (A and C). Migration distances of molecular mass markers are indicated (kDa). The blot was developed with anti-c-myc mAb. The functionality of secreted scFv-based trimerbodies was demonstrated by ELISA against plastic immobilized LM-111 (B and D) and by FACS on CD3+ Jurkat cells and CD3- U937 cells (E). Anti-CD3 (OKT3) and anti-MHC class I (W6/32) mAbs were used as controls.
To investigate whether TIE domains can drive the trimerization of two different scFv, we generated a bispecific N/C-trimerbodyXVIII by fusing the L36 scFv to the N-terminus and the scFv derived from the anti-CD3 OKT3 mAb to the C-terminus of a TIEXVIII domain (L36N/COKT3) (Fig. 1). As a control, we generated an OKT3 scFv C-trimerbodyXVIII (COKT3). Both OKT3 scFv-based trimerbodies were secreted in functional active form by transfected human HEK-293 cells, but yielded significantly lower levels than L36 scFv-based trimerbodies (Fig. 2C). Western blot analysis, under reducing conditions, of conditioned media from HEK-293 cells showed that the migration pattern of the secreted COKT3 and L36N/COKT3 was consistent with the molecular weight calculated from the polypeptide sequences (37.3 and 64.5 kDa, respectively). Functional analysis showed that the bispecific L36N/COKT3 trimerbody bound specifically to both immobilized LM111 [as determined by ELISA (Fig. 2D)], and native CD3 complex expressed on the T cell surface [as determined by flow cytometry (Fig. 2E)]. In contrast, the OKT3 scFv-based C-trimerbodyXVIII bound to the surface of CD3+ cells, but did not interact with LM111 (Fig. 2D and E).
Structural characterization of bispecific scFv-based N/C-terminal trimerbodies
The N-trimerbodyXVIII (L36N) and the bispecific N/C-trimerbodyXVIII (L36N/COKT3) were purified from conditioned medium by immobilized metal affinity chromatography, which yielded proteins that were > 95% pure by reducing SDS-PAGE (Fig. 3A). The functionality of the purified trimerbodies was demonstrated by ELISA and flow cytometry. Figure 3B shows that the dose-dependent binding curves of L36N and L36N/COKT3 to plastic immobilized LM111 were comparable. Furthermore, bispecific L36N/COKT3 trimerbody recognized cell surface CD3 as efficiently as the native mAb OKT3 (mouse IgG2a) (Fig. 3C).

Figure 3. Functional characterization of purified trimerbodies. Reducing SDS-PAGE of purified N-trimerbodyXVIII (L36N) and N/C-trimerbodyXVIII (L36N/COKT3) (A), antigen titration ELISA against plastic immobilized LM-111 (B) and FACS on CD3+ Jurkat cells (C).
The trimeric nature of the molecules was confirmed by SEC-MALLS measurements. The sample of L36N eluted from the size exclusion column as a major symmetric peak at 12.8 mL with a small portion of high molecular weight aggregates eluting at the exclusion volume of the column. The mass calculated from the dispersed light at the center of the peak is 103 kDa, close to the expected mass of 112 kDa for the trimer (Fig. 4A). The sample of L36N/COKT3 produced a similar chromatogram with a larger proportion of aggregates eluting at the exclusion volume and a peak at 11.9 mL with a measured mass of 187 kDa at its center (Fig. 4B). This value is very close to the 196 kDa mass expected for the trimer. Both measured molar masses are, within the experimental error, the same as the calculated values for trimeric molecules, indicating they are indeed trimers in solution.
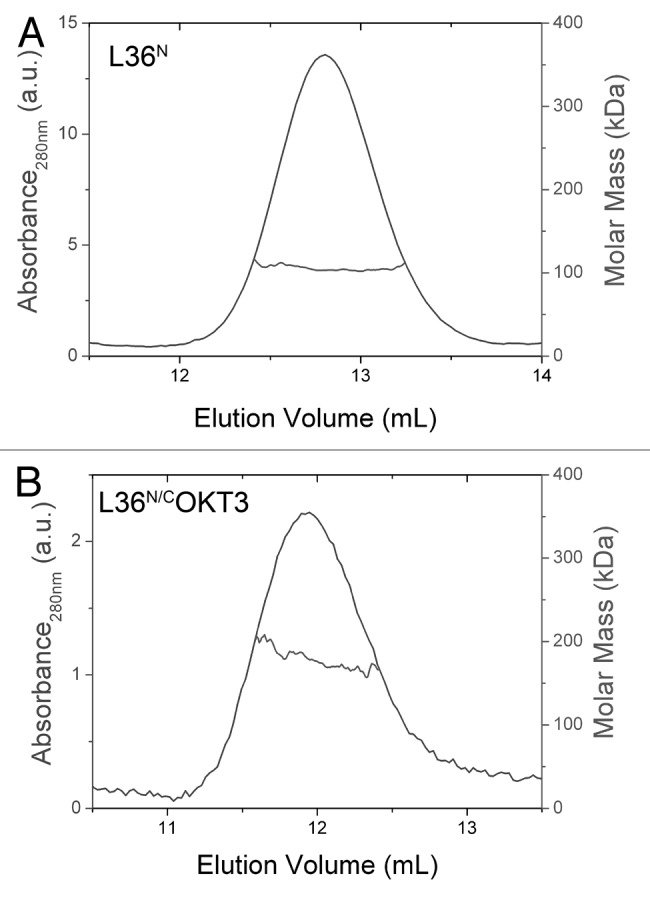
Figure 4. Structural characterization of purified trimerbodies. Oligomeric analysis of the L36N (A) and L36N/COKT3 (B) trimerbodies. Size exclusion chromatogram profile, as measured by UV absorbance at 280 nm (blue trace) and molar mass (red trace), as measured by MALLS (only the data at the central part of the peak is shown, and can be read at the right hand axis, in red color). The mass measured at the center of the peak is indicated.
The serum stability of both L36N and the L36N/COKT3 was studied by incubating each of the purified antibodies in human serum at 37°C, for prolonged periods of time. As shown in Figure 5, the N-trimerbodyXVIII (L36N) and the bispecific N/C-trimerbodyXVIII (L36N/COKT3) had similar stability, retaining more than 50% of the initial laminin-binding activity after 96 h incubation.
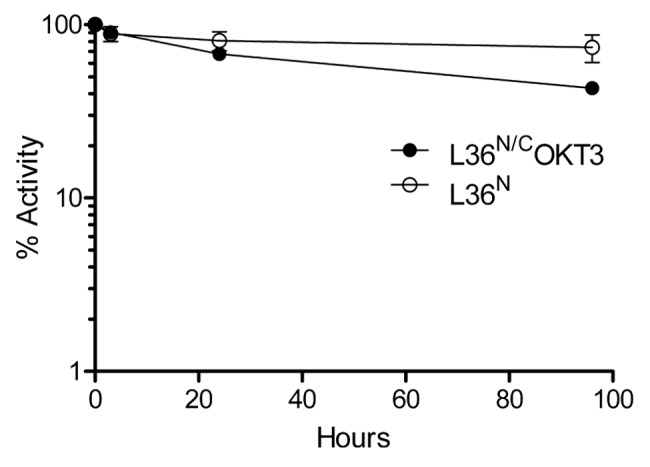
Figure 5. Serum stability of purified of L36N and L36N/COKT3 trimerbodies. ELISA against plastic immobilized LM-111 was performed after incubation 37°C for different time periods in human serum, as explained in material and methods.
Activation of human T cells
To demonstrate the multivalent binding to cell surface receptors, we employed the monospecific anti-CD3 C-trimerbodyXVIII (COKT3) and the bispecific anti-LM111 × anti-CD3 N/C-trimerbodyXVIII (L36N/COKT3). The poor ability of monovalent (Fab and scFv) anti-CD3 antibodies to stimulate T cells compared with bivalent antibodies has long suggested ligand-induced oligomerization as a necessary component of the activation mechanism.18-20 As shown in Figure 6A, Jurkat cells cultured with soluble anti-CD3 mAb OKT3 expressed the activation marker CD69, whereas Jurkat cells incubated in the presence of an isotype control mAb did not express CD69. Jurkat cells cultured with conditioned media from HEK-293 cells transfected with expression vectors encoding different scFv-based trimerbodies (COKT3 or L36N/COKT3), or in the presence of purified L36N/COKT3 trimerbody, upregulated CD69, in a manner similar to the OKT3 mAb. By contrast, Jurkat cells cultured with conditioned media from HEK-293 cells transfected with expression vectors encoding scFv L36-based trimerbodies (L36N, CL36 or L36N/CL36) did not express CD69 (Fig. 6A).

Figure 6. Human T cell activation studies. (A) FACS analysis of CD69 expression by Jurkat cells stimulated either with soluble anti-CD3 (OKT3) or anti-MHC class I (W6/32) mAb, with conditioned media from HEK-293 cells (293-CM) transfected with expression vectors encoding scFv-based trimerbodies (L36N, L36N/CL36, COKT3 or L36N/COKT3), or with purified trimerbodies (L36N or L36N/COKT3). (B) FACS analysis of CD69 expression by Jurkat cells stimulated either with plastic immobilized mAb (imAb), BSA (iBSA) or LM-111 (iLM-111) in the presence of 293-CM from L36N, L36N/CL36, COKT3 or L36N/COKT3 transfected cells or purified L36N or L36N/COKT3 trimerbodies.
To further demonstrate the bispecificity and multivalency of the anti-LM111 x anti-CD3 N/C-trimerbodyXVIII, we performed T cell activation studies in the presence of plastic immobilized BSA (iBSA) or LM111 (iLM111). Purified L36N/COKT3 was added to BSA- or LM111-coated wells and, after washing, the LM111-bound trimerbodies were able to activate Jurkat T cells more effectively than immobilized OKT3 mAb (Fig. 6B). Purified L36N trimerbody was shown to bind to laminin, but the bound trimerbody did not activate human T cells. Similar results were found with conditioned media from HEK-293 cells transfected with expression vectors encoding other scFv-based trimerbodies (Fig. 6B).
Discussion
In this study, we used the trimerbody platform technology for the production of hexavalent scFv-based molecules. We fused scFv antibody fragments with the same or different specificity to both ends of a TIEXVIII domain to produce monospecific or bispecific hexavalent-binding trimerbodies. The monospecific anti-LM111 N-/C-trimerbodyXVIII (L36N/CL36) was generated by fusing the scFv L36 to the N- and C-terminus of a TIEXVIII domain. The bispecific anti-LM111 x anti-CD3 N-/C-trimerbodyXVIII (L36N/COKT3) was similarly generated by fusing the scFv OKT3 onto the C-terminus of a TIEXVIII domain. All the trimerbodies were expressed in functional active form from conditioned medium of transfected HEK293 cells. Variations in expression level were correlated with the scFv clone, rather than with the trimerbody format. Both L36N and L36N/COKT3 molecules were easily purified using standard chromatographic methods.
The purified trimerbodies were trimeric molecules in solution, as unambiguously shown by the light scattering measurements. The elution volumes from the gel filtration column, however, were smaller than those calculated for globular proteins of the same size, according to the calibration of the column with a set of molecular weight markers. This observation is diagnostic of a non-spherical shape of the trimerbodies, which causes them to advance through the column faster than globular molecules of the same size. This behavior is consistent with the design of the trimerbodies and the molecular modeling (Figs. 7 and 8). A similar observation was recently reported for the elongated coiled-coil domain of laminin.21

Figure 7. Modeling of scFv-based trivalent trimerbodies. (A) Superior (left) and lateral (right) views of the anti-LM111 N-terminal trimerbodyXVIII model. (B) Superior (left) and lateral (right) views of the anti-CD3 C-terminal trimerbodyXVIII model. The anti-LM111 scFv is colored in green while the anti-CD3 scFv is colored in blue. In both cases, the N-terminal trimerization region from human collagen XVIII NC1 domain is colored in red. The image shows the different disposition of the linkers according to the terminal side of the trimerization domain. The starting point of the linkers in the N-terminal trimerbodyXVIII model appears completely over the trimerization domain, while in the C-terminal trimerbodyXVIII model, due to the disposition of the last β-strand, the linkers appears to be orientated perpendicular to the trimerization domain.

Figure 8. Modeling of monospecific (A) and bispecific (B) scFv-based hexavalent trimerbodies. (A) Superior (left) and lateral (right) views of a monospecific anti-LM111 N/C-terminal trimerbodyXVIII model. (B) Superior (left) and lateral (right) views of a bispecific anti-LM111 x anti-CD3 N/C-terminal trimerbodyXVIII model. The anti-LM111 scFv domain is colored green while the anti-CD3 scFv is colored in blue. In all cases, the trimerization region of human collagen XVIII NC1 is colored in red. The different configuration between both models shows the putative maximum and minimum aperture that the flexible linkers can achieve. In the monospecific model (A) the aperture of the linker is fixed so that no clashes appear between the initial part of the linker and the trimerization domain. The bispecific model (B) is closed up to the point in which no clashes appear between the scFv domains. In all cases, the linker stability is not affected by the change of orientation.
Monospecific and bispecific hexavalent trimerbodies were very efficient at recognizing antigen either immobilized in plastic, or associated to the cell surface. The dose-dependent binding curves of the monospecific anti-LM111 N-trimerbodyXVIII and the bispecific anti-LM111 x anti-CD3 N-/C-trimerbodyXVIII to plastic immobilized LM111 were comparable. Furthermore, the bispecific L36N/COKT3 trimerbody recognized surface CD3 as efficiently as the native mAb OKT3. The bispecificity of L36N/COKT3 trimerbody was further demonstrated on T cell activation assays. This molecule was very efficient and specific at inducing CD69 expression by human T cells when pre-incubated on plastic-bound LM111, but not on plastic-bound BSA. In this work, we used an extracellular matrix protein (laminin) and a cell surface receptor (CD3) as model antigens to demonstrate the potential and versatility of trimerbody molecules. Although activation of T cells by antibody-mediated CD3 crosslinking provides us with relevant information regarding trimerbody multivalency, it is obvious that multivalent anti-CD3 trimerbodies, at least in its current configuration, cannot be systemically administered due to the risk of unspecific T cell activation.
Although another group has also shown that collagen-derived sequences [in this case a short collagen-like peptide scaffold (Gly-Pro-Pro)10] can promote trimerization of fused scFv antibody fragments,22 we demonstrated here, for the first time, the generation of functional bispecific and multivalent scFv-based antibodies that contain modified versions of the trimerization region of human collagen XVIII.
Since flexibility between antigen binding sites is an important aspect in the design of multivalent antibodies, all the generated trimerbodies have 21 residue flexible linkers allowing diverse binding geometries. Analysis of the trivalent scFv-based N- or C-trimerbody models suggests a tripod-shaped structure with the scFv domains outwardly oriented (Fig. 7). Hexavalent scFv-based trimerbodies are hourglass-shaped molecules (Fig. 8). The 21 residues linker provides a maximum area of influence for each side of the hexavalent trimerbodies around 80 Å of radius. This area of access is slightly higher in the C-terminal side of the hexavalent trimerbodies due to the final orientation of the trimerization region of collagen XVIII NC1, whose last β-strand is oriented almost perpendicular to the plane of trimerization, contrary to the N-terminal side of the trimerization domain, whose final orientation is practically parallel to the plane of trimerization. The trimerbody platform technology has a high degree of structural plasticity and would allow, by adjustment of the linker length, the design of more compact and rigid molecules to meet specific requirements.
Although numerous strategies have been used to generate multivalent antibodies, a number of them have drawbacks due to their immunogenicity, associated unwanted functional effects, poor stability or complex engineering.4 The trimerbody platform has important advantages over conventional strategies, e.g., potentially no immunogenicity, strong association, high expression level and solubility. Furthermore, hexavalent monospecific or bispecific trimerbodies are easy to engineer due to the small size of TIE domains.
The trimerbody platform offers therefore promising opportunities for the development of the next-generation of dual targeting agents. Bispecific antibodies are one of the most promising and exciting areas of protein engineering, and the range of their therapeutic uses have expanded beyond oncology to inflammatory, autoimmune and infectious diseases.3 Bispecific antibodies suitable for therapeutic use can be produced by somatic hybridization, chemical conjugation and genetic engineering.23 Many recombinant bispecific antibodies are symmetric or asymmetric IgG-like molecules (bivalent, trivalent or tetravalent) produced by the dimeric assembly of two identical heavy chains.17 These antibody formats are full-length IgG in which constant domains represent more than 70% of the protein content. A different approach for the generation of recombinant bispecific antibodies is the DNL method that allows the generation of trivalent or hexavalent molecules in which the constant domains also represent a large proportion of the protein content.14 In contrast, the trimerbody platform allows the generation of symmetric hexavalent bispecific molecules with less than 20% of the protein content serving a structural function.
Hexavalent trimerbodies may have applications as effective trapping agents, or for the simultaneous neutralization of different angiogenic factors or disease-modulating cytokines. Other applications include the dual targeting of two receptors for cancer therapy and the development of improved agonistic reagents for immunotherapy. Further interests are the development of fusion proteins for the targeted delivery of a therapeutically active moiety, e.g., angiogenic inhibitors, cytokines, toxins, enzymes.
Material and methods
Reagents and antibodies
The mAbs used included 9E10 (Abcam), anti-c-myc tag, Tetra-His (Qiagen, GmbH) specific for His-tagged proteins, OKT3 (Janssen-Cilag Pty Limited) anti-human CD3ε and phycoerythrin (PE)-conjugated anti-human CD69 mAb (clone FN50, BD Biosciences). The polyclonal antibodies included: horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG, Fc specific, (Sigma-Aldrich), and IRDye800 conjugated donkey anti-mouse IgG (H&L) (Rockland Immunochemicals). Laminin-111 (LM111) extracted from the Engelbreth-Holm-Swarm (EHS) mouse tumor was from Invitrogen Life Technologies. Bovine serum albumin (BSA) was from Sigma-Aldrich.
Cells and culture conditions
HEK-293 (CRL-1573) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza) supplemented with 10% (vol/vol) heat inactivated Fetal Calf Serum (FCS) (Invitrogen Life Technologies), unless otherwise stated. Jurkat clone E6–1 (TIB-152) cells were maintained in RPMI-1640 (Lonza) supplemented with heat-inactivated 10% FCS. All of these cell lines were obtained from the American Type Culture Collection.
Construction of expression vectors
To construct the pCR3.1-L36-hNC1XVIII expression vector, the N-terminal trimerization region from human collagen XVIII NC1 domain (hNC1XVIII) was synthesized by GeneArt AG, and subcloned as NotI/XbaI into the vector pCR3.1-L36,24 containing the L36 (anti-LM111) single-chain Fv (scFv) gene25. To generate a scFv-based C-terminal trimerbody molecule (C-trimerbody) a DNA fragment―coding for the hNC1XVIII, a flexible linker and the VH chain of the L36 scFv―was synthesized by GeneArt AG, and subcloned as ClaI/XhoI into the vector pCR3.1-L36,24 to obtain the pCR3.1-hNC1XVIII-L36 expression vector. To generate an anti-CD3 scFv-based C-trimerbody, a DNA fragment coding for a flexible linker and the OKT3 scFv26 was synthesized by GeneArt AG, and subcloned as BamHI/XbaI into the vector pCR3.1-hNC1XVIII-L36, resulting in pCR3.1-hNC1XVIII-OKT3. To generate a monospecific (anti-LM111) scFv-based N/C-terminal trimerbody the BamHI/XbaI fragment from plasmid pCR3.1-hNC1XVIII-L36 was ligated into the BamHI/XbaI digested backbone of plasmid pCR3.1-L36-hNC1XVIII, to obtain the plasmid pCR3.1-L36-hNC1XVIII-L36. To generate a bispecific (anti-LM111 x anti-CD3ε) scFv-based N/C-terminal trimerbody the BamHI/XbaI fragment from plasmid pCR3.1-hNC1 XVIII-OKT3 was ligated into the BamHI/XbaI digested backbone of plasmid pCR3.1-L36-hNC1XVIII, to obtain the plasmid pCR3.1-L36-hNC XVIII-OKT3.
Expression and purification of recombinant antibodies
Stable cell lines were generated in HEK 293 cells, and the proteins purified from conditioned medium. HEK-293 cells were transfected with the appropriate expression vectors using calcium phosphate,27 and selected in DMEM with 0.5 mg/ml G-418 (Sigma-Aldrich). Supernatants from transiently and stably transfected cell populations were analyzed for protein expression by ELISA, SDS-PAGE and western blotting using anti-myc mAb and IRDye800 conjugated donkey anti-mouse IgG. Stably transfected cell lines were used to collect serum-free conditioned medium that was dialyzed against PBS (pH 7.4) and loaded onto a HisTrap HP 1 ml column using and ÄKTA Prime plus system (GE Healthcare). The purified proteins were dialyzed against PBS and stored at -80°C.
Size exclusion chromatography-multi-angle laser light scattering (SEC-MALLS)
Static light scattering experiments were performed at room temperature using a Superdex 200 Size Exclusion Chromatography column (GE HealthCare) attached in-line to a DAWN-HELEOS light scattering detector (Wyatt Technology Corporation). This column had been previously calibrated with the Gel Filtration Molecular Weight Standards (Bio-Rad Laboratories Ltd.). The column was equilibrated with running buffer (PBS + 0.03% NaN3, 0.1 μm filtered) and the SEC-MALLS system was calibrated with a sample of BSA; at 1 g/L. Then 100 μL samples of the different antibodies at 0.1 mg/mL in PBS were injected into the column at a flow rate of 0.5 mL/min. Data acquisition and analysis were performed using ASTRA software (v 5.3.4.19, Wyatt). Based on numerous measurements on BSA samples at 1 g/L under the same or similar conditions we estimate that the experimental error in the molar mass is around 5%.
ELISA
The ability of N-, C- and N/C-trimerbodies to bind LM111 was studied by ELISA as described.16 Briefly, Maxisorp (NUNC Brand Products) plates were coated with LM111 (0.5 μg/well) and after washing and blocking with 200 μl 5% BSA in PBS, 100 μl with indicated amount of purified protein or supernatant from transiently or stably transfected HEK-293 cells were added for 1 h at room temperature. After three washes, 100 μl of anti-myc mAb (10 μg/ml) were added for 1 h at room temperature. After three washes, 100 μl of HRP-conjugated goat anti-mouse IgG were added for 1 h at room temperature, after which the plate was washed and developed. Antigen titration was performed with serial dilutions of the purified trimerbodies.
Western blotting
Supernatant were separated by reducing SDS-PAGE in 12% Tris-Glycine gels (Bio-Rad Laboratories Ltd.) and transferred using iBlot system (Invitrogen Life Technologies). After blocking with LI-COR blocking solution (LI-COR), proteins were detected with anti-myc mAb and an IRDye800 conjugated donkey anti-mouse IgG. Images were taken using an Odyssey Infrared Imaging system (LI-COR).
Flow cytometry
The ability of N-, C- and N/C-trimerbodies to bind to cell surface antigens (CD3ε) was studied by FACS as described previously.16,28 Briefly, cells were incubated with supernatants or purified trimerbody molecules (10 μg/ml) for 30 min. After washing, the cells were incubated with mAb Tetra-His in 100 μl for 30 min. After washing, the cells were treated with appropriate dilutions of PE-conjugated goat anti-mouse IgG. The samples were analyzed with a Beckman-Coulter FC-500 Analyzer (Beckman-Coulter).
T cell activation assays
To study the ability of N-, C- and N/C-trimerbodies to activate specifically human T cell in solution, Jurkat cells (105) were incubated overnight with supernatants or purified trimerbody molecules (2 μg/ml). For activation in solid phase, supernatants or purified trimerbody molecules (5 μg/ml) were incubated with plastic immobilized LM111 (1 μg/well) in round-bottom 96-well plates (BD Biosciences) for 30 min at 4°C. After washing and blocking, 105 Jurkat cells were added and cultured overnight. After 16 h cells were collected and the surface expression of CD69 examined by flow cytometry.
Serum stability
One microgram of each purified trimerbody was incubated in 60% human serum at 37°C for up to 96 h. Samples were removed for analysis at 3 h, 24 h and 96 h following the start of incubation and frozen until the entire study was completed. As a control, a second set of serum-exposed samples was frozen immediately to represent a zero time point. Aliquots were then subjected to ELISA to test their capability to bind LM111.
Molecular modeling
Each monomer of the trivalent N-trimerbody contains one L36 scFv domain. The scFv domains were built by homology modeling29 using the structure of 2GHW.B30 from the PDB database31 as template. The alignment between the template and the sequence of L36 using blast32 had an e-value of 2e-79 and 70% sequence identity. Each monomer of the trivalent C-trimerbody contains one OKT3 scFv domain. The OKT3 scFv domain was built using as template 1QOK.A,33 with e-value of 3e-81 and 75% of sequence identity. Both monomer types contain the N-terminal trimerization region from human collagen XVIII NC1 domain (hNC1XVIII). The structure of the trimer of hNC1XVIII domains is stored in the PDB with code 3HSH.34 This structure was used to guide the trimerization of the modeled monomers by means of structural superimposition of the NC1 domains with STAMP,35 forming the hexavalent N/C-trimerbody. Each monomer of the hexavalent monospecific N/C-trimerbody contains two identical instances of the L36 scFv antibody fragment, while the monomers of the hexavalent bispecific N/C-trimerbody contain one L36 scFv and one OKT3 scFv.
Acknowledgments
This study was supported by grants from Ministerio de Ciencia e Innovación (BIO2008–03233), Ministerio de Economía y Competitividad (BIO2011–22738), and Comunidad de Madrid (S-BIO-0236–2006 and S2010/BMD-2312) to L.A-V.; from Fondo de Investigación Sanitaria/Instituto de Salud Carlos III (PI08/90856 and PS09/00227) to L.S.; Ministerio de Ciencia e Innovación (BIO2008–00205) to B.O., and CTQ2011–28680 to F.J.B. A.B-T. and A.A-C. were supported by Programa Torres Quevedo from Ministerio de Economía y Competitividad, co-founded by the European Social Fund (PTQ-09–01–01089 and PTQ11–04604, respectively). P.S-V. was supported by a Fundación de Investigación Biomédica-Hospital Universitario Puerta de Hierro training grant. The authors declare no conflict of interest related to this work.
Glossary
Abbreviations:
- DNL
dock-and-lock method
- EHS
Engelbreth-Holm-Swarm mouse tumor
- Fab
fragment antigen binding
- LM111
Laminin-111
- mAb
monoclonal antibody
- NC1
noncollagenous domain
- scFv
single-chain Fv
- SEC-MALLS
Size exclusion chromatography-multi-angle laser light scattering
- TIE
Trimerization
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/22698
References
- 1.Reichert JM. Marketed therapeutic antibodies compendium. MAbs. 2012;4:413–5. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanz L, Cuesta AM, Compte M, Alvarez-Vallina L. Antibody engineering: facing new challenges in cancer therapy. Acta Pharmacol Sin. 2005;26:641–8. doi: 10.1111/j.1745-7254.2005.00135.x. [DOI] [PubMed] [Google Scholar]
- 3.Kontermann R. Dual targeting strategies with bispecific antibodies. MAbs. 2012;4:182–97. doi: 10.4161/mabs.4.2.19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuesta AM, Sainz-Pastor N, Bonet J, Oliva B, Alvarez-Vallina L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010;28:355–62. doi: 10.1016/j.tibtech.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–36. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 6.Pack P, Plückthun A. Miniantibodies: use of amphipathic helices to produce functional, flexibly linked dimeric FV fragments with high avidity in Escherichia coli. Biochemistry. 1992;31:1579–84. doi: 10.1021/bi00121a001. [DOI] [PubMed] [Google Scholar]
- 7.Li S-L, Liang S-J, Guo N, Wu AM, Fujita-Yamaguchi Y. Single-chain antibodies against human insulin-like growth factor I receptor: expression, purification, and effect on tumor growth. Cancer Immunol Immunother. 2000;49:243–52. doi: 10.1007/s002620000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu S, Shively L, Raubitschek A, Sherman M, Williams LE, Wong JYC, et al. Minibody: A novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996;56:3055–61. [PubMed] [Google Scholar]
- 9.Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA. 1993;90:6444–8. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deyev SM, Waibel R, Lebedenko EN, Schubiger AP, Plückthun A. Design of multivalent complexes using the barnase*barstar module. Nat Biotechnol. 2003;21:1486–92. doi: 10.1038/nbt916. [DOI] [PubMed] [Google Scholar]
- 11.Borsi L, Balza E, Carnemolla B, Sassi F, Castellani P, Berndt A, et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood. 2003;102:4384–92. doi: 10.1182/blood-2003-04-1039. [DOI] [PubMed] [Google Scholar]
- 12.Halin C, Gafner V, Villani ME, Borsi L, Berndt A, Kosmehl H, et al. Synergistic therapeutic effects of a tumor targeting antibody fragment, fused to interleukin 12 and to tumor necrosis factor α. Cancer Res. 2003;63:3202–10. [PubMed] [Google Scholar]
- 13.Cloutier SM, Couty S, Terskikh A, Marguerat L, Crivelli V, Pugnières M, et al. Streptabody, a high avidity molecule made by tetramerization of in vivo biotinylated, phage display-selected scFv fragments on streptavidin. Mol Immunol. 2000;37:1067–77. doi: 10.1016/S0161-5890(01)00023-2. [DOI] [PubMed] [Google Scholar]
- 14.Rossi EA, Goldenberg DM, Cardillo TM, McBride WJ, Sharkey RM, Chang C-H. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting. Proc Natl Acad Sci USA. 2006;103:6841–6. doi: 10.1073/pnas.0600982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sánchez-Arévalo Lobo VJ, Cuesta AM, Sanz L, Compte M, García P, Prieto J, et al. Enhanced antiangiogenic therapy with antibody-collagen XVIII NC1 domain fusion proteins engineered to exploit matrix remodeling events. Int J Cancer. 2006;119:455–62. doi: 10.1002/ijc.21851. [DOI] [PubMed] [Google Scholar]
- 16.Cuesta ÁM, Sánchez-Martín D, Sanz L, Bonet J, Compte M, Kremer L, et al. In vivo tumor targeting and imaging with engineered trivalent antibody fragments containing collagen-derived sequences. PLoS ONE. 2009;4:e5381. doi: 10.1371/journal.pone.0005381. 10.1371 /journal.pone.0005381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuesta AM, Sánchez-Martín D, Blanco-Toribio A, Villate M, Enciso-Álvarez K, Alvarez-Cienfuegos A, et al. Improved stability of multivalent antibodies containing the human collagen XV trimerization domain. MAbs. 2012;4:226–32. doi: 10.4161/mabs.4.2.19140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kappler J, Kubo R, Haskins K, White J, Marrack P. The mouse T cell receptor: comparison of MHC-restricted receptors on two T cell hybridomas. Cell. 1983;34:727–37. doi: 10.1016/0092-8674(83)90529-9. [DOI] [PubMed] [Google Scholar]
- 19.Meuer SC, Hodgdon JC, Hussey RE, Protentis JP, Schlossman SF, Reinherz EL. Antigen-like effects of monoclonal antibodies directed at receptors on human T cell clones. J Exp Med. 1983;158:988–93. doi: 10.1084/jem.158.3.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaye J, Gillis S, Mizel SB, Shevach EM, Malek TR, Dinarello CA, et al. Growth of a cloned helper T cell line induced by a monoclonal antibody specific for the antigen receptor: interleukin 1 is required for the expression of receptors for interleukin 2. J Immunol. 1984;133:1339–45. [PubMed] [Google Scholar]
- 21.Santos-Valle P, Guijarro-Muñoz I, Cuesta ÁM, Alonso-Camino V, Villate M, Álvarez-Cienfuegos A, et al. The Heterotrimeric Laminin Coiled-Coil Domain Exerts Anti-Adhesive Effects and Induces a Pro-Invasive Phenotype. PLoS ONE 2012; 7:e39097; 10.1371/ journal.pone.0039097. [DOI] [PMC free article] [PubMed]
- 22.Fan C-Y, Huang C-C, Chiu W-C, Lai C-C, Liou G-G, Li H-C, et al. Production of multivalent protein binders using a self-trimerizing collagen-like peptide scaffold. FASEB J. 2008;22:3795–804. doi: 10.1096/fj.08-111484. [DOI] [PubMed] [Google Scholar]
- 23.Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? MAbs. 2009;1:539–47. doi: 10.4161/mabs.1.6.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanz L, Kristensen P, Blanco B, Facteau S, Russell SJ, Winter G, et al. Single-chain antibody-based gene therapy: inhibition of tumor growth by in situ production of phage-derived human antibody fragments blocking functionally active sites of cell-associated matrices. Gene Ther. 2002;9:1049–53. doi: 10.1038/sj.gt.3301725. [DOI] [PubMed] [Google Scholar]
- 25.Sanz L, García-Bermejo L, Blanco FJ, Kristensen P, Feijóo M, Suárez E, et al. A novel cell binding site in the coiled-coil domain of laminin involved in capillary morphogenesis. EMBO J. 2003;22:1508–17. doi: 10.1093/emboj/cdg150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal antibodies defining distinctive human T cell surface antigens. Science. 1979;206:347–9. doi: 10.1126/science.314668. [DOI] [PubMed] [Google Scholar]
- 27.Compte M, Blanco B, Serrano F, Cuesta AM, Sanz L, Bernad A, et al. Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA x anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther. 2007;14:380–8. doi: 10.1038/sj.cgt.7701021. [DOI] [PubMed] [Google Scholar]
- 28.Alonso-Camino V, Sánchez-Martín D, Compte M, Sanz L, Alvarez-Vallina L. Lymphocyte display: a novel antibody selection platform based on T cell activation. PLoS ONE. 2009;4:e7174. doi: 10.1371/journal.pone.0007174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–91. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 30.Hwang WC, Lin Y, Santelli E, Sui J, Jaroszewski L, Stec B, et al. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J Biol Chem. 2006;281:34610–6. doi: 10.1074/jbc.M603275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boehm MK, Corper AL, Wan T, Sohi MK, Sutton BJ, Thornton JD, et al. Crystal structure of the anti-(carcinoembryonic antigen) single-chain Fv antibody MFE-23 and a model for antigen binding based on intermolecular contacts. Biochem J. 2000;346:519–28. doi: 10.1042/0264-6021:3460519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boudko SP, Sasaki T, Engel J, Lerch TF, Nix J, Chapman MS, et al. Crystal structure of human collagen XVIII trimerization domain: A novel collagen trimerization Fold. J Mol Biol. 2009;392:787–802. doi: 10.1016/j.jmb.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–23. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]