

Abstract
A Biopharmaceutics Drug Disposition Classification System (BDDCS) was proposed to serve as a basis for predicting the importance of transporters in determining drug bioavailability and disposition. BDDCS may be useful in predicting: routes of drug elimination; efflux and absorptive transporters effects on oral absorption; when transporter-enzyme interplay will yield clinically significant effects (e.g. low drug bioavailability and drug-drug interactions); and transporter effects on post-absorptive systemic drug levels following oral and i.v. dosing. For highly soluble, highly permeable Class 1 compounds, metabolism is the major route of elimination and transporter effects on drug bioavailability and hepatic disposition are negligible. In contrast for the poorly permeable Class 3 and 4 compounds, metabolism only plays a minor role in drug elimination. Uptake transporters are major determinants of drug bioavailability for these poorly permeable drugs and both uptake and efflux transporters could be important for drug elimination. Highly permeable, poorly soluble, extensively metabolized Class 2 compounds present the most complicated relationship in defining the impact of transporters due to a marked transporter-enzyme interplay. Uptake transporters are unimportant for Class 2 drug bioavailability, (ensure space after,) but can play a major role in hepatic and renal elimination. Efflux transporters have major effects on drug bioavailability, absorption, metabolism and elimination of Class 2 drugs. It is difficult to accurately characterize drugs in terms of the high permeability criteria, i.e. ≥90% absorbed. We suggest that extensive metabolism may substitute for the high permeability characteristic, and that BDDCS using elimination criteria may provide predictability in characterizing drug disposition profiles for all classes of compounds.
In the early 1990s, our group carried out interaction studies in humans with cyclosporine, tacrolimus and sirolimus with and without ketoconazole, an inhibitor of cytochrome P450 3A (CYP3A) and P-glycoprotein (P-gp), as well as with and without rifampin, an inducer of CYP3A and P-gp [1–6]. These studies suggested that the major effect of the interaction was on bioavailability, as opposed to clearance, and that this interaction occurred primarily in the gut [3]. At that time we asked ourselves three questions: (1) Why does the CYP3A-P-gp exposure interaction appear to be more important in the intestine versus the liver? (2) Why do some CYP3A-efflux transporter substrates exhibit this interplay and others do not? and (3) What about drugs that are not metabolized; how important is transporter-transporter interplay? We believed that if we could answer these questions that this would serve as the basis for predicting drug exposure for a new molecular entity.
Cellular and animal studies from our laboratory over the next decade examining transporter-enzyme interplay led us to make 22 predictions concerning the drug absorption and disposition in our 2005 paper [7]. A number of these predictions, based on the development of a Biopharmaceutics Drug Disposition Classification System (BDDCS), are discussed in this MiniReview. BDDCS is a modification of the FDA’s Biopharmaceutics Classification System (BCS), which was based on the work of Amidon and colleagues [8]. The core idea of the BCS is that an in vitro transport model, centrally embracing permeability and solubility, with qualifications related to pH and dissolution, may qualify drug products for a waiver of in vivo bioequivalence studies. The objective of the BCS is to predict in vivo performance of drug products from in vitro measurements of permeability and solubility. However, we believe that the framework of the BCS can serve the interest of the earliest stages of drug discovery research in predicting the absorption/disposition of new molecular entities.
The BCS categorizes drugs into four classes according to their solubility and permeability as depicted in fig. 1. In 2000, the FDA used the BCS system as a science-based approach to allow waiver of in vivo bioavailability and bioequivalence testing of immediate-release solid dosage forms for Class 1 high solubility, high permeability drugs when such drug products also exhibited rapid dissolution [9]. A drug substance is considered ‘highly soluble’ when the highest dose strength is soluble in 250 ml or less of aqueous media over a pH range of 1–7.5 at 37°C. A drug substance is considered to be ‘highly permeable’ when the extent of absorption (parent drug + metabolites) in humans is determined to be ≥90% of an administered dose, based on a mass balance determination or in comparison to an intravenous reference dose. Note that the BCS definition of permeability, is scientifically confounding in that a rate parameter, permeability is characterized based on an extent measure, that is, how much is absorbed. Thus, in the BCS, a kinetic parameter is defined in terms of a thermodynamic measure.
Figure 1.
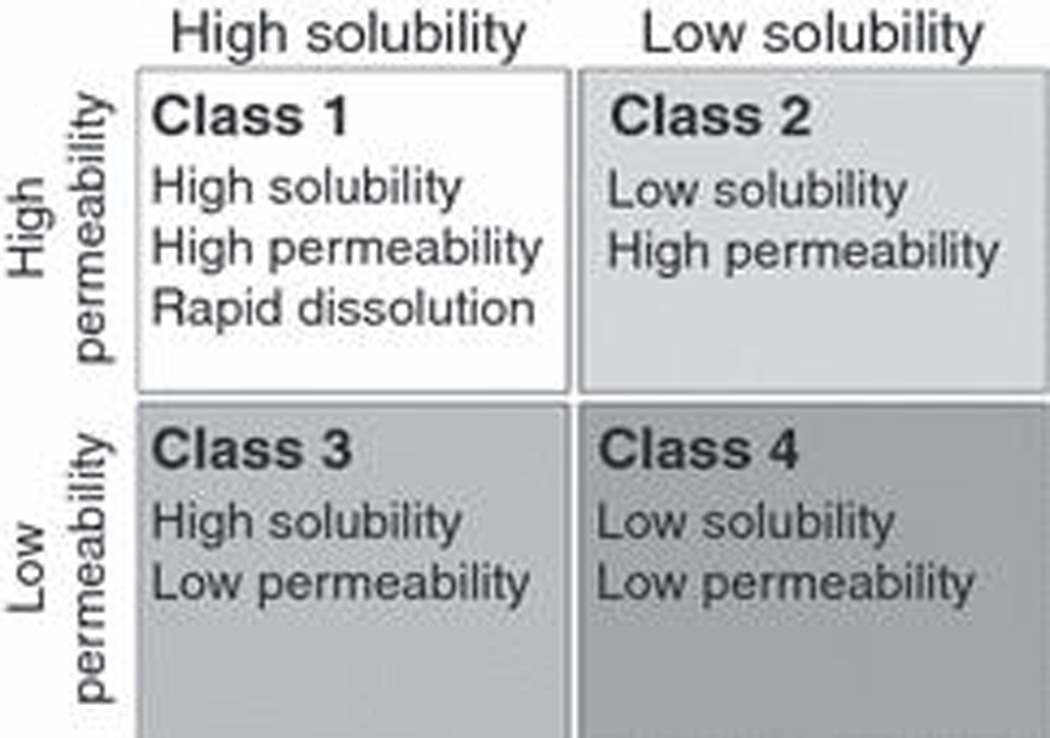
The Biopharmaceutics Classification System (BCS) as proposed by Amidon et al. [8] and promulgated by the FDA [9].
Initially, based on a limited number (34) of compounds for which human in vivo intestinal permeability measures were experimentally determined, the correlation between permeability rate and extent of absorption held reasonably well. But that is no longer true. The FDA has classified as ‘highly permeable’ a number of drugs where absorption is ≥90% in humans but the permeability of these compounds is less than that for metoprolol (the drug designated in the BCS as the cut off permeability for highly permeable compounds). A number of these drugs showing high extent of absorption but poor permeability have been recently tabulated [10] and include cefadroxil, cephradine, levofloxacin, loracarbef, ofloxacin, pregabalin and sotalol.
I have no argument with the FDA setting the criteria for a waiver of in vivo bioequivalence being ≥90% absorbed in humans and can fully support waivers based on high solubility and high extent of absorption. But I am concerned when a science-based regulatory Agency, such as the FDA, redefines a well-recognized scientific parameter such as permeability in terms of a regulatory definition that is not consistent with the scientific criteria. The basis of my concern is that such a regulatory redefinition hampers scientific progress and the development of predictive approaches, as we have recently outlined [11]. We are already seeing the negative effects of this redefinition as investigators attempt to use in vitro measures of permeability to try to correctly predict BCS Class 1 drugs. The consequences of this redefinition should be of particular relevance to programmes whose objective is to develop predictive pharmacokinetic/pharmacodynamic drug metabolism and toxicity for new molecular entities, such as the COST B25 initiative.
Wu and Benet [7] reviewed 130 compounds that had been classified in the literature into the four BCS categories. They recognized that the major route of elimination for the high permeability Class 1 and Class 2 drugs in humans was metabolism, while the major route of elimination for the poorly permeable Class 3 and Class 4 drugs in humans was renal and biliary excretion of unchanged drug. Since, at least for drugs on the market, the extent of metabolism is better characterized than the extent of absorption, Wu and Benet proposed that in the BDDCS, drugs be categorized in terms of the extent of metabolism and solubility versus permeability and solubility [7]. This immediately eliminated the situation where drugs were classified in more than one class because of the uncertainty of permeability measures from study to study, since at least for drugs on the market the extent of metabolism is much better characterized than the extent of absorption. The implications from the BDDCS for a new molecular entity is that if a surrogate measure of intestinal absorption is available, such as permeability through a Caco-2 cellular system, it would be possible to predict the major route of elimination for this new molecular entity in humans. Thus, Benet and Wu proposed the BDDCS as shown in fig. 2 with ≥70% metabolism being the cut off for extensive metabolism [7]. They also noted that there were relatively few drugs where the extent of metabolism was between 30% and 70% and that most drugs are either very highly metabolized or very poorly metabolized.
Figure 2.
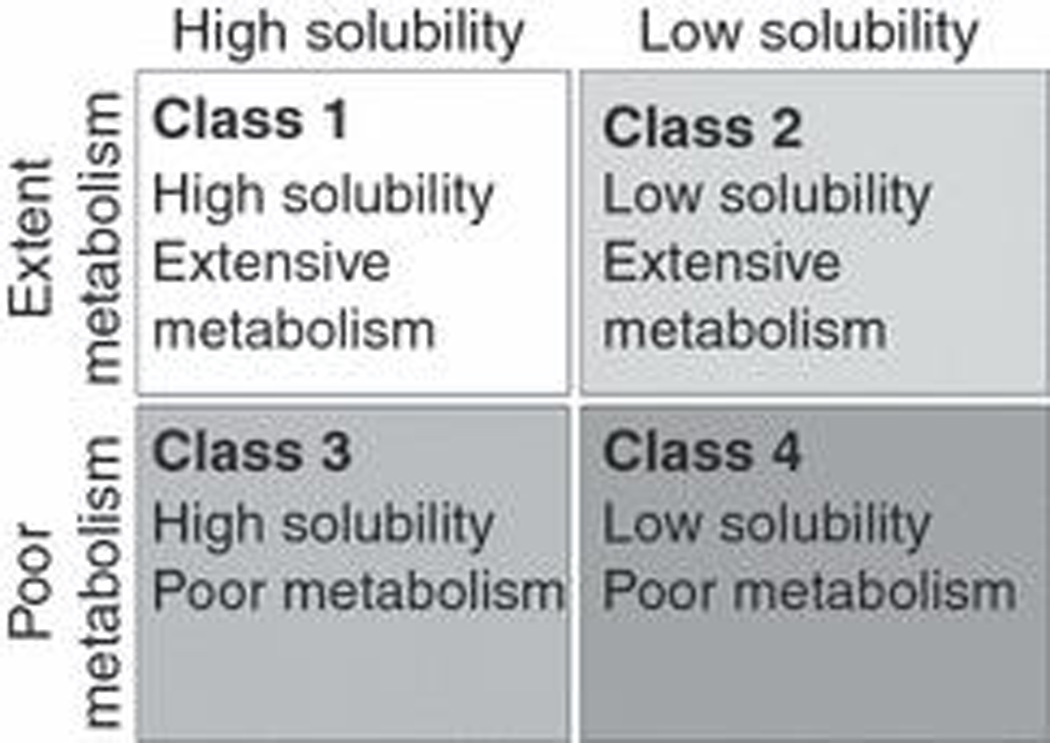
The Biopharmaceutics Drug Disposition Classification System (BDDCS) as proposed by Wu and Benet [7].
Benet and coworkers [12] recognized that for 29 drugs for which measured human intestinal permeabilities were available, the extent of metabolism correctly predicted high versus low permeability for 27 of 29 or 93% of the laboriously and expensively measured human intestinal permeabilities in terms of BCS. Thus, Benet and colleagues recommended ‘that regulatory agencies add the extent of drug metabolism (i.e. ≥90% metabolized) as an alternate method for the extent of drug absorption (i.e. ≥90% absorbed) in defining Class 1 drugs suitable for a waiver of in vivo studies of bioequivalence’. The authors proposed that the following criteria be used to define ≥90% metabolized for marketed drugs: ‘Following a single oral dose to humans, administered at the highest dose strength, mass balance of the Phase I oxidative and Phase II conjugative drug metabolites in the urine and faeces measured either as unlabeled, radioactive labelled or non-radioactive labelled substances, account for ≥90% of the drug dosed. This is the strictest definition for a waiver based on metabolism. For an orally administered drug to be ≥90% metabolized by Phase I oxidative and Phase II conjugative processes, it is obvious that the drug must be absorbed’.
It should be noted that for such a simple categorization as BCS, permeability does a remarkable job in predicting both the extent of absorption and the extent of metabolism. However, for oral drugs that reach the market the metabolism prediction is somewhat better, probably because oral drugs do not reach the market unless they do have acceptable absorption, while there are no restraints on extent of metabolism [11]. BDDCS may not be sufficient for the regulatory agencies, but the July 2008 EMEA Draft Revised ‘Guideline on the Investigation of Bioequivalence’ includes metabolism as a criterion for permeability [13]. However, BDDCS was not developed to allow Class 1 biowaivers; the intent of BDDCS was to give scientists and clinicians a roadmap for predicting drug disposition and drug-drug interaction characteristics very early, with little additional expense.
Fig. 3 summarizes the predictions from BDDCS related to the effects of transporters in the gut and liver following oral dosing of drugs [7]. For Class 1, highly soluble-highly permeable-extensively metabolized, drugs transporter effects will be minimal in the intestine and the liver. Even compounds like verapamil that can be shown in certain cellular systems (e.g. MDR1-MDCK) to be a substrate of P-gp will exhibit no clinically significant P-gp substrate effects in the gut and the liver. Thus, a major proposition (and possibly the primary advance in knowledge) of BDDCS is that Class 1 drugs are not substrates for transporters in the intestine and the liver. However, BDDCS predictions only apply to the intestine and the liver since Class 1 drugs will be substrates for transporters at the blood brain barrier and in the kidney.
Figure 3.
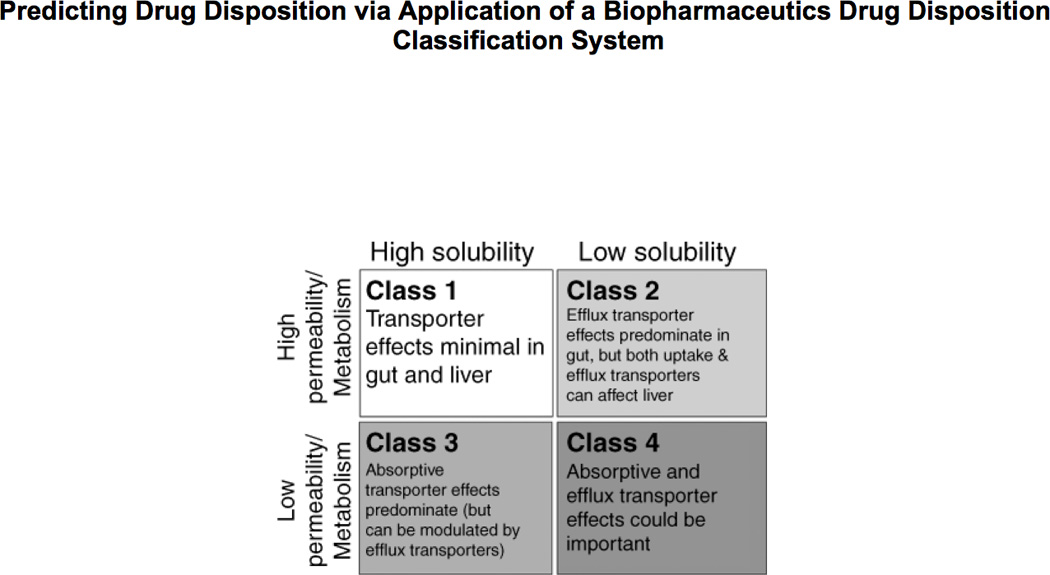
Transporter effects predicted by BDDCS following oral dosing.
From fig. 3, it is seen that for Class 2 drugs, efflux transporter effects will predominate in the intestine. The high permeability of these compounds will allow ready access into the gut membranes, but the low solubility will limit the concentrations coming into the enterocytes, thereby preventing saturation of the efflux transporters. Transporter-enzyme interplay will be primarily important for Class 2 compounds that are substrates for CYP3A and Phase II gut enzymes (e.g. glucuronosyltransferases), where efflux transporter effects can control the access of the drug to the gut enzymes. The cartoon in fig. 4 depicts our conception of the interaction between CYP3A and P-gp in the intestine. Three drug molecules are depicted. These molecules all represent the same drug and are only differentiated by their outcome. Drug is absorbed by passive processes into the enterocyte where it may be metabolized by the enzyme. However, the drug is also subject to active efflux back into the intestine, thereby allowing further access to the enzyme on subsequent passive absorption. The open circle molecule enters the enterocyte, is not metabolized by CYP3A or effluxed back into the lumen by P-gp. This molecule then proceeds into the hepatic portal vein and to the liver. The solid circle molecule is absorbed into the enterocyte and is metabolized to the open square product upon its first encounter with the enzyme. The metabolite either passes into the hepatic portal blood or back into the gut lumen. However, the grey diamond molecule is absorbed; it is not metabolized by the enzyme; it is effluxed back into the gut lumen by P-gp and this cycling occurs on two further occasions, where upon the fourth entry into the enterocyte the diamond molecule is metabolized to the open square metabolite. The cartoon in fig. 4 depicts the transporter as controlling the access of the drug to the enzyme, giving the enzyme multiple opportunities to prevent the intact drug molecule from entering the blood stream. Thus, we hypothesize that the enzyme and the transporter are working in a coordinated manner as a protective process to keep foreign substances out of the body. Note that if no P-gp were present, or if the P-gp were inhibited, the enzyme would only have one opportunity to metabolize the drug as it diffuses through the enterocyte (as for the open circle molecule). However, when the P-gp is present, or to an even greater extent when it is induced, the enzyme has multiple opportunities to metabolize the drug. We recognized, based on fig. 4, that intestinal metabolism of a drug could be changed as a function of P-gp activity without either inhibiting or inducing intestinal enzymes. That is, if efflux by P-glycoprotein is inhibited, the drug molecule passes through the intestine in a single pass and intestinal metabolism will decrease since there is less access of drug to the enzymes.
Figure 4.

Cartoon depicting CYP3A4 and P-gp interplay in the enterocytes.
The left hand side of fig. 5 depicts the process detailed above with respect to an apical intestinal dose of drug as was represented in fig. 4. The right hand side of fig. 5 depicts the interplay between CYP3A and P-gp if the drug was dosed on the basolateral side of fig. 4. Under these conditions, the drug molecule encounters the enzyme prior to coming in contact with the efflux transporters. Thus, substrates diffusing into cells will be pumped out by P-gp but not diffuse back in because it is against the concentration gradient. Under such a condition, less metabolites would be formed and more parent would traverse the membrane. On the right hand side of fig. 5, it is obvious that inhibiting P-gp will maintain the drug within the cell leading to enhanced metabolism, while up-regulating P-gp will cause drug to sweep through the cell leading to less metabolism. We recognized that the left hand side of fig. 5 represented the interplay of CYP3A and P-gp that would occur in the intestine, where the efflux transporter is encountered prior to the enzyme, while the right hand side of fig. 5 depicts the CYP3A and P-gp interplay in the liver where drug molecules enter the liver on the basolateral side, then encounter the enzyme prior to being effluxed by P-gp into the bile.
Figure 5.
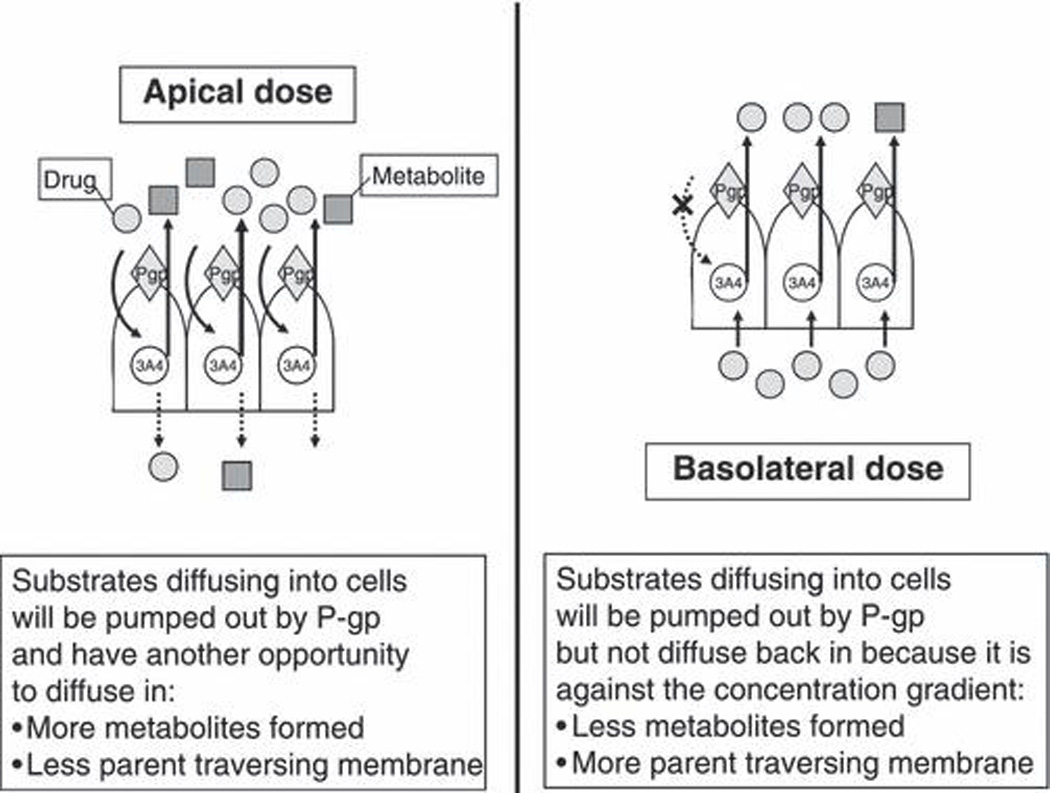
Cartoon depicting differential effects of CYP3A4 and P-gp interplay following apical and basolateral dosing.
In the late 1990s, a series of studies mostly in cellular systems and in animals confirmed the hypothesized CYP3A and P-gp interplay depicted in fig. 4, demonstrating that inhibition or induction of intestinal CYP3A and/or P-gp had marked effects on the bioavailability of a number of drugs as summarized by Zhang and Benet [14]. The interplay in the liver as depicted on the right side of fig. 5 was confirmed in studies of tacrolimus using the isolated perfused rat liver [15] and in vivo rat studies with digoxin [16].
Recognizing the differential effects between the CYP3A and P-gp interplay in the intestine versus the liver allowed us to predict changes in exposure resulting from drug interactions of each of these proteins or the concomitant effect of both as shown in table 1 [17]. In the late 1990s, a number of drugs were removed from the market when drug interactions following oral dosing led to significantly greater drug interactions than were predicted from in vitro enzyme inhibition studies. In the late 1990s, it was recognized that inhibition of CYP3A in the intestine and the liver would lead to increased exposure for drugs such as cisapride, terfenadine, mebefradil and astemizole, as was noted in the approved labels for these compounds. However, it was not generally recognized at that time that there is a marked overlap between compounds that inhibit the CYP3A and those that inhibit P-gp, although this was first noted in 1995 [18]. Thus, as depicted in table 1, inhibition of CYP3A and P-gp in the gut led to an additive, or possibly synergistic, increase in area under the curve (AUC) following oral dosing, which would not be predicted from in vitro microsomal studies. Even in vivo intravenous studies would not predict the extent of the interaction due to the opposing effects in the liver, where inhibition of CYP3A increases AUC but inhibition of P-gp decreases AUC, as explained in fig. 5. Today, we well recognize the interplay of efflux transporters and gut enzyme in terms of drug interactions and we will not make the mistakes of the late 1990s, when these potential, serious, toxic interactive effects occurred.
Table 1.
Predicted exposure (area under the curve) changes for in vivo – in situ studies.
| Gut | Liver | |
|---|---|---|
| Inhibit P-gp | ⇑ | ⇓ |
| Inhibit 3A | ⇑ | ⇑ |
| Inhibit P-gp + 3A | ⇑⇑ | ⇔⇑⇓ |
Returning to fig. 3, note that the BDDCS predicts that both uptake and efflux transporters can affect drug disposition in the liver. Thus, inhibition or induction of uptake hepatic transporters such as the OATPs, OATs and OCTs can lead to changes in hepatic metabolism even when hepatic enzymes are unaffected. We first demonstrated this for digoxin in rats [16] and more recently in a series of studies concerning the disposition of atorvastatin and its two active hydroxyl metabolites in cellular, perfused rat liver, whole rat and humans studies [19–21].
In fig. 3, BDDCS predicts that for Class 3 highly soluble-poorly permeable-poorly metabolized drugs, uptake transporters will be important for intestinal absorption and liver entry for these poorly permeable compounds. However, once these poorly permeable drugs get into the enterocyte or the hepatocyte, efflux transporter effects can also occur. Reflect on the drugs that Professors Yuichi Sugiyama and Richard Kim study in double transfected cellular systems, i.e. both uptake and efflux transporters added (e.g. fexofenadine [22], telmisartan [23] and rosuvastatin [24]). Reflect on the compounds showing improved absorption via a transporter prodrug effect (e.g. PEPT1) such as acyclovir [25] and gemcitabine [26]. All of these substrates are non-metabolized Class 3 and Class 4 drugs that require an uptake transporter in the intestine to achieve effective systemic concentrations.
Finally, BDDCS allows potential drug-drug interactions to be predicted [7,17]. For Class 1 drugs, only metabolic interactions need to be considered in the intestine and the liver. For Class 2 drugs metabolic, efflux transporter and efflux transporter-enzyme interplay in the intestine must be considered. While in the liver, metabolic, uptake transporter, efflux transporter and transporter-enzyme interplay can occur. For Class 3 and Class 4 drugs, uptake transporter, efflux transporter and uptake-efflux transporter interplay will be of major importance.
Conclusions
Understanding transporter-enzyme interactions in terms of the permeability and solubility of drug compounds offers the potential for predicting: (1) major routes of elimination as shown in for the BDDCS system; (2) transporter effects on drug absorption (see 2009 review entitled, ‘The role of transporters in the pharmacokinetics of orally administered drugs’ [27]); (3) the prediction of food effects (not covered here but reviewed in [28]); (4) transporter effects on post-absorption systemic levels and after i.v. dosing; (5) enzyme-transporter interplay; and (6) drug-drug interaction potential and its relationship to enzyme-transporter interplay.
Acknowledgements
Preparation of this MiniReview and the studies in the author’s laboratory were supported in part by NIH grants GM 75900 and GM 61390, and by an unrestricted grant from Amgen. The author is appreciative of the significant assistance of Ms. Frances Peterson in the preparation of this manuscript.
References
- 1.Hebert MF, Roberts JP, Prueksaritanont T, Benet LZ. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin Pharmacol Ther. 1992;52:453–457. doi: 10.1038/clpt.1992.171. [DOI] [PubMed] [Google Scholar]
- 2.Gomez DY, Wacher VJ, Tomlanovich SJ, Hebert MF, Benet LZ. The effects of ketoconazole on the intestinal metabolism and bioavailability of cyclosporine. Clin Pharmacol Ther. 1995;58:15–19. doi: 10.1016/0009-9236(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 3.Wu C-Y, Benet LZ, Hebert MF, Gupta SK, Rowland M, Gomez DY, et al. Differentiation of absorption and first-pass gut and hepatic metabolism in humans: studies with cyclosporine. Clin Pharmacol Ther. 1995;58:492–497. doi: 10.1016/0009-9236(95)90168-X. [DOI] [PubMed] [Google Scholar]
- 4.Floren LC, Bekersky I, Benet LZ, Mekki Q, Dressler D, Lee JW, et al. Tacrolimus oral bioavailability doubles with coadministration of ketoconazole. Clin Pharmacol Ther. 1997;62:41–49. doi: 10.1016/S0009-9236(97)90150-8. [DOI] [PubMed] [Google Scholar]
- 5.Hebert MF, Fisher RM, Marsh CL, Dressler D, Berkersky I. Effects of rifampin on tacrolimus pharmacokinetics in healthy volunteers. J Clin Pharmacol. 1999;39:91–96. doi: 10.1177/00912709922007499. [DOI] [PubMed] [Google Scholar]
- 6.Floren LC, Christians U, Zimmerman JJ, Neefe L, Schorer R, Rushworth D, et al. Sirolimus oral bioavailability increases ten-fold with concomitant ketoconazole. Clin Pharmacol Ther. 1999;65:159. [Google Scholar]
- 7.Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a Biopharmaceutics Drug Disposition Classification System. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 8.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 9.Food and Drug Administration. Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Rockville, MD: Food and Drug Administration; 2000. Retrieved from http://www.fda.gov/cder/guidance/index.htm. [Google Scholar]
- 10.Chen ML, Yu L. The use of drug metabolism for prediction of intestinal permeability (dagger) Mol Pharmaceut. 2009;6:74–81. doi: 10.1021/mp8001864. [DOI] [PubMed] [Google Scholar]
- 11.Larregieu CA, Benet LZ. Use of drug metabolism and Caco-2 transport for prediction of intestinal permeability revisited using accepted scientific definitions vs an FDA surrogate. Mol Pharmaceut. 2009 (In press) [Google Scholar]
- 12.Benet LZ, Amidon GL, Barends DM, Lennernäs H, Polli JE, Shah VP, et al. The use of BDDCS in classifying the permeability of marketed Drugs. Pharm Res. 2008;52:483–488. doi: 10.1007/s11095-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.European Medicines Agency. Guideline on the investigation of bioequivalence CPMP/WEP/QWP/1401/98. European Medicines Agency Web site (July 2008) 2008 Retrieved from http://www.emea.europa.eu/pdfs/human/qwp/140198enrev1.pdf.
- 14.Zhang Y, Benet LZ. The gut as a barrier to drug absorption. Clin Pharmacokinet. 2001;40:159–168. doi: 10.2165/00003088-200140030-00002. [DOI] [PubMed] [Google Scholar]
- 15.Wu C-Y, Benet LZ. Disposition of tacrolimus in isolated perfused rat liver: influence of troleandomycin, cyclosporine, and GG918. Drug Metab Dispos. 2003;31:1292–1295. doi: 10.1124/dmd.31.11.1292. [DOI] [PubMed] [Google Scholar]
- 16.Lau YY, Wu C-Y, Okochi H, Benet LZ. Ex situ inhibition of hepatic uptake and efflux significantly changes metabolism: hepatic enzyme-transporter interplay. J Pharmacol Exp Ther. 2004;308:1040–1045. doi: 10.1124/jpet.103.061770. [DOI] [PubMed] [Google Scholar]
- 17.Benet LZ, Cummins CL, Wu C-Y. Transporter-enzyme interactions: implications for predicting drug-drug interactions from in vitro data. Curr Drug Metab. 2003;4:393–398. doi: 10.2174/1389200033489389. [DOI] [PubMed] [Google Scholar]
- 18.Wacher VJ, Wu C-Y, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog. 1995;13:129–134. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- 19.Lau YY, Okochi H, Huang Y, Benet LZ. Multiple transporters affect the disposition of atorvastatin and its two active hydroxy metabolites: application of in vitro and ex situ systems. J Pharmacol Exp Ther. 2006;316:762–771. doi: 10.1124/jpet.105.093088. [DOI] [PubMed] [Google Scholar]
- 20.Lau YY, Okochi H, Huang Y, Benet LZ. Pharmacokinetics of atorvastatin and its hydroxy metabolites in rats and the effects of concomitant rifampicin single doses: relevance of first-pass effect from hepatic uptake transporters, and intestinal and hepatic metabolism. Drug Metab Dispos. 2006;34:1175–1181. doi: 10.1124/dmd.105.009076. [DOI] [PubMed] [Google Scholar]
- 21.Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81:194–204. doi: 10.1038/sj.clpt.6100038. [DOI] [PubMed] [Google Scholar]
- 22.Cvetkovic M, Leake B, Fromm MF, Wilkinson GR, Kim RB. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab Dispos. 1999;27:866–871. [PubMed] [Google Scholar]
- 23.Ishiguro N, Maeda K, Saito A, Kishimoto W, Matsushima S, Ebner T, et al. Establishment of a set of double transfectants coexpressing organic anion transporting polypeptide 1B3 and hepatic efflux transporters for the characterization of the hepatobiliary transport of telmisartan acylglucuronide. Drug Metab Dispos. 2008;36:796–805. doi: 10.1124/dmd.107.018903. [DOI] [PubMed] [Google Scholar]
- 24.Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008;36:2014–2023. doi: 10.1124/dmd.108.021410. [DOI] [PubMed] [Google Scholar]
- 25.Han H, De Vrueh RL, Rhie JK, Covitz KM, Smith PL, Lee CP, et al. 5′-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm Res. 1998;15:1154–1159. doi: 10.1023/a:1011919319810. [DOI] [PubMed] [Google Scholar]
- 26.Song X, Lorenzi PL, Landowski CP, Vig BS, Hilfinger JM, Amidon GL. Amino acid ester prodrugs of the anticancer agent gemcitabine: synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport. Mol Pharmaceut. 2005;2:157–167. doi: 10.1021/mp049888e. [DOI] [PubMed] [Google Scholar]
- 27.Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26:2039–2054. doi: 10.1007/s11095-009-9924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Custodio JM, Wu C-Y, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]