

Abstract
STAT1 exists in phosphorylated (pSTAT1) and unphosphorylated (uSTAT1) forms each regulated by IFN-γ. Although STAT1 is a key mediator of the IFN-γ signaling pathway, an essential component of the host cancer immunosurveillance system, STAT1 is also overexpressed in certain human cancers where the functions of pSTAT1 and uSTAT1 are ill-defined. Using a murine model of soft tissue sarcoma (STS), we demonstrate that disruption of the IFN effector molecule IRF8 decreases pSTAT1 and increases uSTAT1 in STS cells, thereby increasing their metastatic potential. We determined that the IRF8 gene promoter was hypermethylated frequently in human STS. An analysis of 123 human STS specimens revealed that high uSTAT1 levels in tumor cells was correlated with a reduction in disease-specific survival, whereas high pSTAT1 levels in tumor cells was correlated with an increase in disease-specific survival. In addition, uSTAT1 levels were negatively correlated with pSTAT1 levels in these STS specimens. Mechanistic investigations revealed that IRF8 suppressed STAT1 transcription by binding the STAT1 promoter. RNAi-mediated silencing of STAT1 in STS cells was sufficient to increase expression of the apoptotic mediators Fas and Bad and to elevate the sensitivity of STS cells to Fas-mediated apoptosis. Together, our findings show how the phosphorylation status of pSTAT1 determines its function as a tumor suppressor, with uSTAT1 acting as a tumor promoter that acts by elevating resistance to Fas-mediated apoptosis to promote immune escape.
Introduction
Signal transducer and activator of transcription 1 (STAT1) is a DNA-binding protein that mediates IFN-γ-dependent gene expression. IFN-γ is a pleiotropic cytokine produced by activated T lymphocytes and natural killer (NK) cells. The IFN-γ/STAT1 signaling pathway was originally identified to play an essential role in host defense against microbial and viral infections. Recent studies, however, revealed that the IFN-γ/STAT1 signaling pathway is also critically important in host suppression of tumor development (1-3). Mice lacking the p53 tumor suppressor gene and STAT1 formed a wider spectrum of tumors compared with STAT1 wt mice lacking only p53 (4). Furthermore, STAT1−/− mice formed significantly more carcinogen-induced sarcomas than wt mice. Therefore, STAT1 was viewed as a tumor suppressor (1). On the other hand, the IFN-γ/STAT1 signaling pathway is a major signaling pathway for modulating both pro- and anti-inflammatory responses, and approximately 20% of human cancers are estimated to develop from chronic inflammation (5). Indeed, it has recently been reported that chronic IFN-γ signaling leads to hyperactivation of STAT1 and chronic inflammation-mediated spontaneous colorectal cancer development (6). In addition, STAT1 accumulation and hyperactivation have been observed in multiple types of cancers (3, 7-10). Therefore, the IFN-γ/STAT1 signaling pathway might be a double-edged sword that functions to either suppress or promote cancer development depending on the cellular context.
Binding of IFN-γ to its physiological receptor, the IFN-γR, induces oligomerization of receptors, followed by rapid activation of the receptor-associated JAKs by trans-phosphorylation. The activated JAKs phosphorylate the intracellular domain of the IFN-γR, which then serves as a docking site for STAT1. The cytoplasmic STAT1 (cSTAT1) exists primarily in an unphosphorylated state (uSTAT1). uSTAT1 is recruited to the phosphorylated IFN-γR and is then itself phosphorylated. The phosphorylated STAT1s (pSTAT1) form homodimers and translocate to the nucleus to regulate gene expression by binding to gamma activation site (GAS) elements (TTCN2-4 G/TAAA) in the promoters of IFN-γ target genes. Therefore, pSTAT1 is the key mediator of the IFN-γ signaling pathway. In contrast, uSTAT1 predominately exists in the cytoplasm and is constitutively expressed in most cells. However, pSTAT1 also induces STAT1 expression to increase uSTAT1 accumulation in the cytoplasm. In response to IFN-γ, STAT1 is rapidly phosphorylated but only lasts a few hours, whereas uSTAT1, newly synthesized by pSTAT1-activated transcription, can persist for several days (11).
The function of pSTAT1 in regulation of IFN-γ target genes is well-established. uSTAT1 was originally considered as the latent form of STAT1. However, it has since been demonstrated that uSTAT1, in the absence of IFN-γ stimulation, also functions as a transcription factor (12, 13). These observations indicate that both pSTAT1 and uSTAT1 are transcription factors and may both be involved in the IFN-γ-mediated tumor suppression or promotion. However, the relative role of pSTAT1 and uSTAT1 in tumor development is not well-defined. In a previous study, we showed that disruption of IRF8 function resulted in a dramatic up-regulation of uSTAT1 in mouse soft tissue sarcoma cells (14). Here, we examined cSTAT1/uSTAT1 and nuclear STAT1 (nSTAT1/pSTAT1) protein levels in 123 human STS specimens and observed that cSTAT1 level is inversely correlated with disease-specific survival, whereas pSTAT1 is positively correlated with disease-specific survival. Furthermore, we mechanistically demonstrated that uSTAT1 is a negative regulator of the Fas-mediated apoptosis pathway that suppresses FasL-induced apoptosis at least partially through repressing Fas and Bad expression in sarcoma cells. Our data thus suggest that uSTAT1 is a tumor promoter and pSTAT1 is a tumor suppressor in human STS.
Materials and Methods
Mice and cells
BALB/c (H-2d) mice were obtained from the National Cancer Institute (NCI, Frederick, MD). All mice were housed, maintained and studied in accordance with approved NIH and Georgia Health Sciences University guidelines for animal use and handling. CMS4 cell line has been characterized as previously described (15).
Human STS tumor specimens
Human STS specimens were collected from surgical specimens at The University of Texas M. D. Anderson Cancer Center and formatted into a tissue microarray (TMA) as previous described (16). Specifically, one hundred twenty-three archived paraffin blocks from STS surgical specimens were utilized For TMA construction (Supplemental Table 1), H&E-stained sections were reviewed from each of these STS tumor blocks by a sarcoma pathologist (AJL) to define areas of homogeneous, viable tumor. Using an automated TMA apparatus (ATA-27, Beecher Instruments), 0.6 mm punch samples were obtained from donor AS blocks. The selected tissue cores were formatted into a standard 4.5 × 2 × 1 cm recipient block. Two tissue cores were taken for each case and one TMA block containing a total of 246 cores of STS tumor tissue was constructed. Sections (4 μm) were cut, and one standard H&E stained slide was examined to verify the presence of viable tumor.
Immunohistochemistry
Immunohistochemical staining was performed at the Georgia Pathology Service using anti-STAT1 mAb (BD Biosciences). The specimens were blocked with normal serum (1.5%) and then incubated in primary antibody for 30 min, followed by rinsing and staining with appropriate anti-species biotinylated antibody (1:2,000) for another 30 min. Color was developed by incubation with 3,3′-diaminobenzidine solution (Sigma), followed by rinsing and counterstaining with hematoxylin. Positive and negative controls were run in parallel. Labeling intensity was graded by 2 separate observers (AJL and GL) in both the cytoplasmic and nuclear compartments as none (=0), weak (=1), moderate (=2), or strong (=3); the percentage of positive tumor cells (distribution) was estimated from the 2 paired TMA samples for each case as previously described (16). Anti-CD8 antibody was obtained from Dako and used to stain individual slides as previously described (17).
Experimental lung metastasis mouse model
The experimental lung metastasis mouse model was carried out as previously described (18).
Analysis of tumor-infiltrating T cells
CMS4 cells were injected into BALB/c mice i.v., and tumor-bearing lungs were digested with collagenase as previously described (18). Cells were stained with FITC-conjugated CD8 mAb and PE-conjugated FasL mAb (Biolegend) and analyzed by flow cytometry.
Apoptosis assay
Cells were stained with Propidium Iodide (PI) and Alex Fluor 647-conjugated Annexin V (Biolegend), and analyzed with flow cytometry.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were carried out using anti-IRF8 antibody (Santa Cruz) as previously described (19). The STAT1 promoter DNA was detected by PCR using mouse STAT1 promoter-specific primers (Supplemental Table 2).
Immunoprecipitation
Immunoprecipation was carried out with anti-Bcl2, Bcl-xL, and Bad mAbs (BD Biosciences) and agarose-protein A beads (Millipore). The immunoprecipitated proteins were blotted and probed with respective antibodies. Signals were detected using goat anti-mouse IgG (Fc specific)-peroxidase (Sigma) and ECL Plus kit (GE Health Care).
Gene silencing
Cells were transiently transfected with scramble siRNA (Dharmacon, Lafayette, CO) and mouse STAT1-specific siRNA (Santa Cruz), respectively. Silencing efficiency was analyzed approximately 24 h after transfection.
DNA methylation analysis: Genomic
DNA was extracted from human high grade unclassified pleomorphic sarcoma/malignant fibrous histocytoma (UPS/MFH) specimens acquired per a University of Texas MD Anderson Cancer Center (UTMDACC) IRB approved protocol. DNA was modified with the CpGenome Universal DNA Modification Kit (Millipore). MS-PCR was performed as previously described (20).
RT-PCR analysis
Total RNA was isolated from cells using Trizol (Invitrogen, San Diego, CA) according to the manufacturer’s instructions, and used for the first strand cDNA synthesis using the MMLV reverse transcriptase (Promega). The cDNA was then used as template for PCR amplification. The sequences of primers are listed in Supplemental Table 2.
Cell surface Fas analysis
Cells were stained with FITC-conjugated anti-mouse Fas mAb (Biolegend) and analyzed by flow cytometry. Fas protein level is quantified by mean fluorescence intensity.
Western blot analysis
Western blotting analysis was performed essentially as previously described (19). The blot was probed with pSTAT1-, STAT1-, Bcl-2-, Bcl-xL-, Bad- and Jak1-specific antibodies (BD biosciences). Anti-β-actin antibody was obtained from Sigma.
Statistical Analysis
All statistical analysis was performed using SAS 9.2 and statistical significance was assessed using an alpha level of 0.05. Patient demographic and clinical characteristics were summarized using medians or proportions as applicable. Survival was estimated using the Kaplan-Meier method with 95% confidence intervals, with disease-specific mortalit as an endpoint. The time to disease-related death was computed from the date of surgery at MD Anderson Cancer Center to the date when the event of death was recorded, or the event was censored at the date of last follow-up assessment in event-free patients. Kaplan-Meier curves were used to determine DSS time; Log-rank testing was used to compare DSS between patient subgroups. Fisher’s exact test was used to assess the difference in proportions between subgroups of patients and associations between markers or between marker and disease status. All computations were carried out in SPSS version 17.
Results
uSTAT1 level is positively correlated with the metastatic potential
Mouse STS cell line CMS4 was stably transfected with the control vector (CMS4.pcDNA), the IRF8 mutants with a point mutation (K to E) at amino acid 79 (CMS4.K79E), and a point mutation (R to E) at amino acid 289 (CMS4.R289E), respectively. CMS4.pcDNA and CMS4.K79E cells were injected i.v. into syngeneic BALB/c mice. Examination of mouse lungs revealed that disruption of IRF8 function significantly increased the metastatic potential of CMS4 cells (Fig. 1A). Western blotting analysis revealed that disruption of IRF8 function significantly increases STAT1 protein level in CMS4 cells (Fig. 1B).
Figure 1. cSTAT1 expression level is positively correlated with sarcoma cell metastatic potential in an experimental metastasis mouse model.
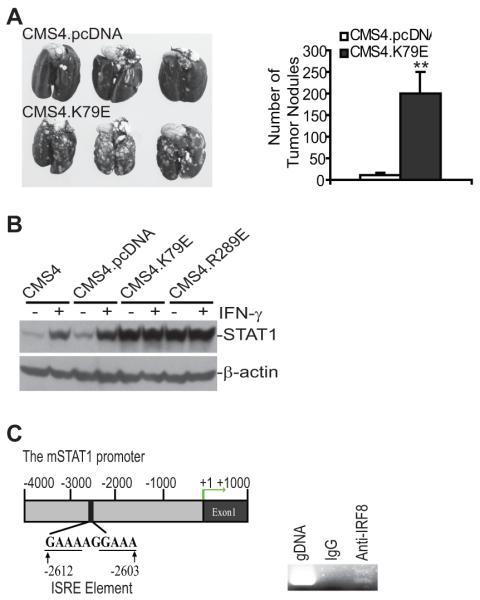
A. Disruption of IRF8 function increased sarcoma cell metastatic potential. CMS4.pCDNA and CMS4.K79E cells (2×105 cells/mouse) were injected into syngeneic mice i.v. and mouse lungs were examined for tumor growth 14 days later. The tumor nodule number in lungs from individual mice were numerated and presented in the right panel. ** p<0.01. B. Disruption of IRF8 function resulted in increased cSTAT1 expression. The indicated 4 cell sublines were cultured in the presence and absence of IFN-γ for 24 h. Cytosol fractions were prepared and analyzed for cSTAT1 protein level by Western blotting analysis. C. ChIP analysis of IRF8 association with the STAT1 promoter chromatin. Left panel: the mouse STAT1 promoter structure showing the ISRE element location. Right panel: ChIP assay was performed with IgG or IRF8-specific antibody to show IRF8 binding to the STAT1 promoter region in CMS4 cells.
Analysis of the mouse STAT1 promoter DNA sequence identified an interferon-stimulated response element (ISRE) (Fig. 1C). ChIP analysis with IRF8-specific antibody demonstrated that IRF8 is associated with the STAT1 promoter chromatin in CMS4 cells (Fig. 1C). Our data thus suggest a positive correlation between STAT1 protein level and STS metastatic potential.
STAT1 expression level is associated with disease-specific survival in human STS
To determine whether the above observations can be extended to human STS specimens we utilized a clinically annotated STS TMA and stained it for STAT1. Ninety-two of the STS tumors (75%) included in the TMA were high grade, the majority (80%) were classified as undifferentiated pleomorphic STS. Positive cytoplasmic (cSTAT1) and nuclear STAT1 (nSTAT1) immunostainings were observed in 116 (94.3%) and 94 (76.4%) tumors, respectively. Cytoplasmic levels were high in 29.3% (n=36) of evaluable tumor samples; high nuclear expression was observed in 15.3% (n=19) of STS tumors (Fig. 2).
Figure 2. STAT1 immunohistochemistry on human sarcoma.

Human STS TMA slides were stained with STAT1 mAb as described in the Materials and Methods. Shown are examples of low (A, C, E, G & I: 40X) and high (B, D & J: 200X; F & H: 400x) power STAT1 staining of strong (A,B) and weak (C,D) cytoplasmic staining, strong (E,F) and weak (G,H) nuclear staining, and negative (I,J) staiing in tumor cells. Infiltrating T-cell lymphocytes serve as a positive internal control (highlighted in panel I & J) and there identity was confirmed using CD3 immunohistochemistry.
Expression intensity of nSTAT1 level was found to be associated with a clear trend towards prolonged disease-specific survival (DSS), however it was not statistically significant; median DSS of STS patients with low nSTAT1 levels was 58 months and has not been reached in the cohort of patients expressing high nSTAT1 levels (p=0.08; Fig. 3A). In contrast, our data demonstrated that high cSTAT1 expression levels were strongly associated with decreased rates of STS-specific survival; median DSS of patients with high cSTAT1 expression levels was 46 months and has not been reached in the low cSTAT1 group (p=0.03; Fig. 3B). In addition, cSTAT1 level was found to be negatively correlation of nSTAT1 level in these STS specimens (p=0.015) with 66.7% of specimens with low cSTAT1 expression scored as high nSTAT1 vs. 92% of specimens with high cSTAT1 expression. In summary, these results imply that nSTAT1 may function as a tumor suppressor, whereas, cSTAT1 as a tumor promoter in STS.
Figure 3. Correlation analysis between STAT1 protein level in tumor cells and disease-specific survival.

A. Kaplan-Meier survival curve for disease-specific survival (DSS) between nSTAT1-positive and negative tumors. STS specimens from 123 STS patients were stained with STAT1-specific mAb and examined for nSTAT1 protein in the tumor cells. The nSTAT1-positive and negative groups were then analyzed for correlation with DSS. B. Kaplan-Meier survival curve for DSS and correlation with cSTAT1 protein level in the tumor cells. STS specimens were analyzed as in A and cSTAT1 high protein level group was compared to the cSTAT1 low protein level group for correlation with DSS. C. The IRF8 promoter DNA is hypermethylated in human STS. Genomic DNA was isolated from dissected human STS cells and treated with bisulfite. The modified DNA was then analyzed with MS-PCR primers specific for the human IRF8 promoter. U: unmethylation; M: methylation.
IRF8 promoter DNA is hypermethylated in human STS
Because STAT1 expression is potentially regulated by IRF8 (Fig. 1), and the IRF8 promoter DNA is frequently methylated in human cancer cells (20-22), we next sought to determine the methylation status of the IRF8 promoter DNA in human STS cells. Genomic DNA was isolated from 10 human STS specimens and the IRF8 promoter DNA methylation status was analyzed. MS-PCR analysis indicated that the IRF8 promoter DNA is hypermethylated in all 10 specimens examined (Fig. 3C).
STAT1 confers sarcoma cell resistance to FasL-induced apoptosis
Acquisition of resistance to apoptosis, including Fas-mediated apoptosis, is often associated with tumor progression (23, 24). The positive correlation between STAT1 level and increased tumor burden (Figs. 1-3) suggest that STAT1 might promote tumor development through regulating apoptosis in sarcoma cells. To test this hypothesis, we silenced uSTAT1 expression in CMS4 cells (Fig. 4A) and analyzed the tumor cell sensitivity to FasL-induced apoptosis. While CMS4 cells were not sensitive to FasL-induced apoptosis, silencing uSTAT1 dramatically increased tumor cell sensitivity to FasL-induced apoptosis in vitro (Fig. 4B) (p<0.01).
Figure 4. Silencing STAT1 expression increases sarcoma cell sensitivity to Fas-mediated apoptosis in vitro.
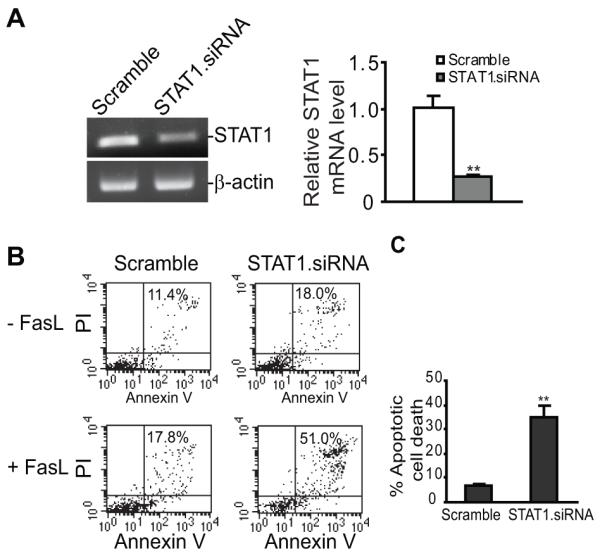
A. CMS4 cells were transfected with scramble and mouse STAT1-specific siRNAs, respectively, overnight, and analyzed for STAT1 expression level by semi-quantitative RT-PCR (left panel) and real-time RT-PCR (right panel). B. The STAT1 siRNA-transfected cells were incubated in the absence or presence of FasL (250 ng/ml) overnight and then stained with PI and Annexin V. The stained cells were analyzed with flow cytometry. The number in each plot indicated the PI and Annexin V double positive cells. Apoptotic cell death was quantified by the formula: percentage of PI+ and Annexin V+ cells in the presence of FasL - percentage of PI+ and Annexin V+ cells without FasL treatment, and presented at the right panel.
uSTAT1 regulates Fas and Bad expression
It has been shown that uSTAT1 directly regulates the expression of several genes, including Bcl-xL, Bcl-2l1, ccnd1, cMyc and STAT3 in several types of cells (25-29). RT-PCR analysis indicated that silencing uSTAT1 expression up-regulates Bcl-xL expression (Fig. 5A). However, the expression levels of Bcl-2l1, ccnd1, cMyc and STAT3 were not changed in STAT1-silenced cells (Fig. 5A), suggesting cell type-dependent regulation of gene expression by uSTAT1. Because STAT1 mediates sarcoma cell sensitivity to Fas-mediated apoptosis (Fig. 4B), we then examined the expression levels of genes with known function in the Fas-mediated apoptosis pathway. RT-PCR analysis revealed that the expression levels of Fas and Bad are up-regulated in STAT1-silenced sarcoma cells (Fig. 5B & C). Consistent with increased Fas mRNA level, STAT1-silenced CMS4 cells also expressed higher level of cell surface Fas protein as compared to the control cells (Fig. 5B). It is known that it is the ratio of expression levels of pro- and anti-apoptotic genes that determines the sensitivity of cells to apoptosis induction. The observation that silencing STAT1 increases Bcl-xL expression level and at the same time significantly increased the tumor cell sensitivity to Fas-mediated apoptosis suggest that Bcl-xL function is neutralized by increased pro-apoptotic protein. To test this hypothesis, we performed immunoprecipitation using Bcl-xL- and Bcl-2-specific mAbs and analyzed Bcl-xL- and Bcl-2-associated pro-apoptotic proteins. We detected that Bad is associated with Bcl-xL protein (Fgi. 5D). Therefore, Bcl-xL and Bad form a protein complex in the CMS4 tumor cells.
Figure 5. STAT1 regulates Bcl-xL, Fas and Bad expression.

A-C. CMS4 cells were transfected with scramble and mouse STAT1-specific siRNAs, respectively, overnight, and analyzed for the expression level of genes as indicated by semi-quantitative RT-PCR. Cell surface Fas protein levels were measured by staining cells with Fas-specific mAb and analyzed by flow cytometry. Fas protein level is quantified by mean fluorescence intensity and presented in bottom of panel C. *p=0.05. D. Bcl-xL and Bad form a protein complex in sarcoma cells. Cell lysates were prepared from CMS4 cells, immunoprecipitated with Bcl-2- and Bcl-xL-specific mAbs, respectively, and then analyzed for Bcl-2, Bcl-xL and Bad protein association by Western blotting analysis.
Tumor-infiltrating cytotoxic T lymphocytes express FasL
Fas is a death receptor that by itself does not initiate apoptosis in tumor cells. FasL is the physiological ligand of Fas. FasL is expressed on the surface of activated cytotoxic T lymphocytes (CTL) (30-32). We reasoned that if STAT1 mediates tumor cell sensitivity to Fas-mediated apoptosis in vivo (Fig. 4), then the tumor-infiltrating CTLs (2) might be the source of FasL that initiates tumor cell apoptosis in vivo. Next, we analyzed human STS specimens for CTL levels. Immunohistochemical staining of tumor specimens with CD8-specific mAb indicated that CTLs are present in 5 of the 5 specimens analyzed. The CTL infiltration level differs from specimens to specimens. However, CTLs are relatively evenly distributed inside the tumor tissues (Fig. 6A). To determine the FasL level in the STS tumor tissue, we injected CMS4 cells into mice i.v. and tumor-bearing lungs were analyzed for CTLs. CTLs consist of approximately 3% of the total cells in the tumor-bearing lung tissues and approximately 22% of these tumor-infiltrating CTLs are FasL+ (Fig. 6B).
Figure 6. Tumor-infiltrating CTLs express FasL.
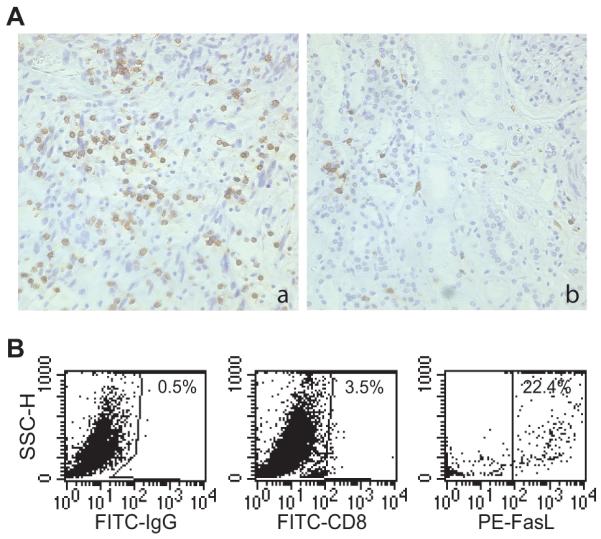
A. Presence of tumor-infiltrating CD8+ T cells in human STS tissues. Human STS specimens were stained with CD8-specific mAb and analyzed for CD8+ T cell level. Shown are representative images of tumor tissues with high (a) and low (b) level of CD8+ T cell infiltration. CD8-specific staining is shown as the brown color, whereas cells that are unreactive are indicated by the blue counterstain. B. Tumor-infiltrating CD8+ T cells express FasL. CMS4 cells were injected i.v. into syngeneic mice. Tumor-bearing lungs were collected and digested with collagenase 14 days later to make single cell suspension. The cell suspension was stained with FITC-conjugated CD8 mAb and PE-conjugated FasL mAb. CD8+ cells were then gated and analyzed for the level of FasL+ cells. The number in each plot indicates the percentage of the specific cell population.
uSTAT1 represses Jak1 expression to inhibit IFN-γ-induced STAT1 phosphorylation
Disruption of IRF8 function not only leads to up-regulation of uSTAT1 (Fig. 1), but also inhibition of IFN-γ-induced STAT1 phosphorylation (Fig. 7A). Analysis of pSTAT1 activity using EMSA indicated that pSTAT1 binding to GAS-containing DNA is also diminished in sarcoma cells overexpressing IRF8 mutants (Fig. 7B). To determine whether diminished STAT1 phosphorylation is due to uSTAT1 up-regulation, we silenced uSTAT1 in CMS4 sarcoma cells and analyzed the expression level of Jak1. RT-PCR analysis revealed that silencing STAT1 in CMS4 cells resulted in up-regulation of Jak1 (Fig. 7C). Our data thus suggest that uSTAT1 is a repressor of Jak1 expression.
Figure 7. uSTAT1 represses Jak1 expression to inhibit IFN-γ-induced STAT1 phosphorylation.

A. Disruption of IRF8 function results in diminished pSTAT1 and Jak1 level in sarcoma cells. The indicated 4 cell sublines were cultured in the presence and absence of IFN-γ for 24 h and analyzed for pSTAT1 and Jak1 protein levels by Western blotting analysis. B. Disruption of IRF8 function results in diminished pSTAT1 activity in sarcoma cells. CMS4 cells were cultured in the presence or absence of IFN-γ for 4 h. Nuclear extracts were prepared from the cells and incubated with a GAS element-containing DNA probe. The protein-DNA interactions were then analyzed by EMSA. C. STAT1 is a repressor of Jak1. CMS4 cells were transfected with scramble and mouse STAT1-specific siRNAs, respectively, overnight and analyzed for STAT1 and Jak1 expression level by semi-quantitative RT-PCR. D. IFN-γ-induced STAT1 activation (pSTAT1) and expression (uSTAT1) kinetics in sarcoma cells. CMS4 cells were cultured in the presence of IFN-γ for the indicated time and analyzed for pSTAT1 and total STAT1 protein level by Western blotting analysis.
Next, we analyzed pSTAT1 and uSTAT1 activation and expression kinetics in CMS4 cells after IFN-γ treatment. As reported in the literature in other types of cells (11), IFN-γ induced rapid STAT1 phosphorylation, however, pSTAT1 also disappeared rapidly (Fig. 7D). In contrast, uSTAT1 started to accumulate after pSTAT1 degradation and showed constant up-regulation for at least 2 days in CMS4 cells after IFN-γ treatment (Fig. 7D). Taken together, our data suggest that uSTAT1 is a repressor of Jak1 and functions as a feedback terminator of the IFN-γ/pSTAT1 signaling pathway.
Discussion
IFN-γ is a pleiotropic cytokine secreted by activated T cells and NK cells and plays a critical role in the host cancer immunosurveillance (1, 33). STAT1 is the key mediator of the IFN-γ signaling pathway and has been shown to function as a tumor suppressor (1, 2, 4). However, STAT1 hyperactivation and chronic IFN-γ signaling is observed to be linked to promotion of inflammation-mediated spontaneous cancer development (6), and STAT1−/− mice are partially protected from leukemia development (34). Furthermore, overexpression of STAT1 is associated with tumor cell resistance to chemotherapeutic agents and radiation (35-45). These studies suggest that STAT1 might also function as a tumor promoter.
In this study, we observed two interesting phenomena in an experimental metastasis mouse sarcoma model in vivo: 1) a positive correlation between uSTAT1 expression level and metastatic potential of mouse sarcoma cells (Fig. 1A); and 2) an inverse correlation between IFN-γ-induced pSTAT1 level and metastatic potential of mouse sarcoma cells (Fig. 7A & B). More interestingly, consistent with what was observed in the mouse sarcoma tumor model (Fig. 7A & B) and in human colorectal and squamous cell cancer patients (2, 3), we observed that human STS patients with high percentage of p/nSTAT1+ tumor cells show a trend of better disease-specific survival (Fig. 3A), whereas STS patients with higher level of cSTAT1 protein in their tumor cells exhibit significantly worse disease-specific survival (Fig. 3B). Our data thereby suggest that pSTAT1 functions as a tumor suppressor and uSTAT1 acts as a tumor promoter in sarcoma cells.
The mechanisms underlying STAT1 in tumor promotion are still elusive. The observation that overexpression of STAT1 is associated with resistance of tumor cells to chemotherapeutic agents and radiation suggest that STAT1 might confer tumor cell resistance to apoptosis to promote tumor development (35-38, 40-45). Indeed, silencing STAT1 enhanced tumor cell sensitivity to apoptosis induction by chemotherapeutic agents and radiation (36, 43, 45). In this study, we demonstrated that silencing STAT1 significantly increased sensitivity of sarcoma cells to FasL-induced apoptosis (Fig. 4). The Fas-FasL system plays a critical role in suppression of tumor development under physiological conditions (46-49). FasL is primarily expressed on activated CTLs (30-32). In this study, we also observed CTL infiltration in human STS tissues (Fig. 6A) and that tumor-infiltrating CTLs express FasL in vivo (Fig. 6B). These results thus suggest that uSTAT1 might confer sarcoma cell resistance to Fas-mediated apoptosis to escapes CTL-mediated tumor suppression in vivo (Figs. 1-3).
uSTAT1 is a transcription factor (50) and has been shown to directly regulate the expression of several genes with known functions in apoptosis regulation, including Bcl-xL and STAT3 (25, 26). We observed that uSTAT1 functions as a repressor of Bcl-xL in sarcoma cells (Fig. 5). However, uSTAT1 also functions as a Bad suppressor (Fig. 5c), and Bad forms a complex with Bcl-xL in sarcoma cells (Fig. 5D). Therefore, uSTAT1-mediated suppression of Bcl-xL does not lead to increased sensitivity of the sarcoma cells to apoptosis induction. Silencing STAT1 resulted in up-regulation of Fas (Fig. 5B), suggesting that uSTAT1 is a Fas repressor, and therefore, uSTAT1 might use repressing Fas expression to confer sarcoma cell resistance to FasL-induced apoptosis.
Jak1 is the key kinase that phosphorylates the IFN-γR and subsequently STAT1. It has been shown that tumor cells use down-regulation of Jak1 to impair the IFN-γ signaling pathway. In this study, we observed that uSTAT1 also functions as a repressor of Jak1 to impair pSTAT1 formation (Fig. 7A & B). Therefore, uSTAT1 not only represses expression of Fas and Bad to confer the tumor cell resistance to Fas-mediated apoptosis (Fig. 4 & 5), but also represses Jak1 expression to potentially terminate STAT1 phosphorylation (Fig. 7). Because IFN-γ/pSTAT1 can up-regulate Fas expression and enhances tumor cell sensitivity to apoptosis induction (39), repression of Jak1 expression by uSTAT1 thus ensures that uSTAT1-mediated Fas repression and apoptosis resistance may not be reversed by IFN-γ signaling in the tumor microenvironment.
Based on the literature and our above observations, we propose a model to illustrate the crosstalk network of pSTAT1 and uSTAT1 in IFN-γ signaling, apoptosis and tumor development (Supplemental Fig. 1). We propose that exposure of sarcoma cells to IFN-γ in the tumor microenvironment induces rapid Jak1 phosphorylation and pSTAT1 formation. pSTAT1 transcriptionally activates Fas and IRF8. uSTAT1 is repressed by IRF8 and cannot be up-regulated by pSTAT1 at the early stage of IFN-γ signaling. This acute IFN-γ signaling results in tumor cell sensitivity to Fas-mediated apoptosis and tumor suppression by FasL+ tumor-infiltrating CTLs. However, IFN-γ-induced pSTAT1 and IRF8 only have a short half-life and uSTAT1 starts to accumulate after pSTAT1 and IRF8 start to degrade (Fig. 7D). uSTAT1 suppresses Fas to confer tumor cell apoptosis resistance. uSTAT1 also represses Jak1 to terminate the IFN-γ signaling in a feedback inhibition manner. Therefore, chronic elevation of uSTAT1 may impair the IFN-γ signaling pathway and thus keep Fas and Bad at a low expression level to maintain tumor cells an apoptosis-resistant phenotype to escape from immunosurveillance to promote tumor development (Supplemental Fig. 1).
Supplementary Material
Acknowledgement
We thank Ms. Kimberly K. Smith for her excellent technical assistance in the immunohistochemical staining of tumor specimens.
Grant Support: National Institutes of Health (CA133085 to K.L), the American Cancer Society (RSG-09-209-01-TBG to K.L.).
Footnotes
Conflict of interest disclosure: None
References
- 1.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 2.Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59:926–33. doi: 10.1136/gut.2009.194472. [DOI] [PubMed] [Google Scholar]
- 3.Laimer K, Spizzo G, Obrist P, Gastl G, Brunhuber T, Schafer G, et al. STAT1 activation in squamous cell cancer of the oral cavity: a potential predictive marker of response to adjuvant chemotherapy. Cancer. 2007;110:326–33. doi: 10.1002/cncr.22813. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–61. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 6.Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203:1391–7. doi: 10.1084/jem.20060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajkumar T, Sabitha K, Vijayalakshmi N, Shirley S, Bose MV, Gopal G, et al. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenwood C, Metodieva G, Al-Janabi K, Lausen B, Alldridge L, Leng L, et al. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J Proteomics. 2011 doi: 10.1016/j.jprot.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 9.Duarte CW, Willey CD, Zhi D, Cui X, Harris JJ, Vaughan LK, et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS One. 2012;7:e29653. doi: 10.1371/journal.pone.0029653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727–38. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehtonen A, Matikainen S, Julkunen I. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J Immunol. 1997;159:794–803. [PubMed] [Google Scholar]
- 12.Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278:1630–2. doi: 10.1126/science.278.5343.1630. [DOI] [PubMed] [Google Scholar]
- 13.Chatterjee-Kishore M, Kishore R, Hicklin DJ, Marincola FM, Ferrone S. Different requirements for signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 in the regulation of low molecular mass polypeptide 2 and transporter associated with antigen processing 1 gene expression. J Biol Chem. 1998;273:16177–83. doi: 10.1074/jbc.273.26.16177. [DOI] [PubMed] [Google Scholar]
- 14.Yang D, Thangaraju M, Browning DD, Dong Z, Korchin B, Lev DC, et al. IFN Regulatory Factor 8 Mediates Apoptosis in Nonhemopoietic Tumor Cells via Regulation of Fas Expression. J Immunol. 2007;179:4775–82. doi: 10.4049/jimmunol.179.7.4775. [DOI] [PubMed] [Google Scholar]
- 15.DeLeo AB, Shiku H, Takahashi T, John M, Old LJ. Cell surface antigens of chemically induced sarcomas of the mouse. I. Mouse leukemia virus related antigens and alloantigens on cultured fibroblasts and sarcomas: description of a unique antigen on BALB/c Meth A sarcoma. J Exp Med. 1977;146:720–34. doi: 10.1084/jem.146.3.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lahat G, Tuvin D, Wei C, Wang WL, Pollock RE, Anaya DA, et al. Molecular prognosticators of complex karyotype soft tissue sarcoma outcome: a tissue microarray-based study. Ann Oncol. 2010;21:1112–20. doi: 10.1093/annonc/mdp459. [DOI] [PubMed] [Google Scholar]
- 17.Liu F, Hu X, Zimmerman M, Waller J, Wu P, Hayes-Jordan A, et al. TNFα Cooperates with IFN-γ to Repress Bcl-xL Expression to Sensitize Metastatic Colon Carcinoma Cells to TRAIL-mediated Apoptosis. PLoS ONE. 2011;6:e16241. doi: 10.1371/journal.pone.0016241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang D, Stewart TJ, Smith KK, Georgi D, Abrams SI, Liu K. Downregulation of IFN-gammaR in association with loss of Fas function is linked to tumor progression. Int J Cancer. 2008;122:350–62. doi: 10.1002/ijc.23090. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerman M, Yang D, Hu X, Liu F, Singh N, Browning D, et al. IFN-γ Upregulates Survivin and Ifi202 Expression to Induce Survival and Proliferation of Tumor-Specific T Cells. PLoS One. 2010;5:e14076. doi: 10.1371/journal.pone.0014076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu X, Yang D, Zimmerman M, Liu F, Yang J, Kannan S, et al. IRF8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer Res. 2011;71:2882–91. doi: 10.1158/0008-5472.CAN-10-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGough JM, Yang D, Huang S, Georgi D, Hewitt SM, Rocken C, et al. DNA methylation represses IFN-gamma-induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol Cancer Res. 2008;6:1841–51. doi: 10.1158/1541-7786.MCR-08-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, Ren S, Howell P, Fodstad O, Riker AI. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008;21:545–58. doi: 10.1111/j.1755-148X.2008.00484.x. [DOI] [PubMed] [Google Scholar]
- 23.Villa-Morales M, Fernandez-Piqueras J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:85–101. doi: 10.1517/14728222.2011.628937. [DOI] [PubMed] [Google Scholar]
- 24.Liu K. Role of apoptosis resistance in immune evasion and metastasis of colorectal cancer. World J Gastrointest Oncol. 2010;2:399–406. doi: 10.4251/wjgo.v2.i11.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111–22. doi: 10.1093/emboj/19.15.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T. Signals through gp130 upregulate bcl-x gene expression via STAT1-binding cis-element in cardiac myocytes. J Clin Invest. 1997;99:2898–905. doi: 10.1172/JCI119484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 28.Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, et al. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and - independent pathways. EMBO J. 2000;19:263–72. doi: 10.1093/emboj/19.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–92. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- 30.Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J Immunol. 2009;182:4217–25. doi: 10.4049/jimmunol.0802793. [DOI] [PubMed] [Google Scholar]
- 31.Kilinc MO, Rowswell-Turner RB, Gu T, Virtuoso LP, Egilmez NK. Activated CD8+ T-effector/memory cells eliminate CD4+ CD25+ Foxp3+ T-suppressor cells from tumors via FasL mediated apoptosis. J Immunol. 2009;183:7656–60. doi: 10.4049/jimmunol.0902625. [DOI] [PubMed] [Google Scholar]
- 32.Fritzsching B, Oberle N, Eberhardt N, Quick S, Haas J, Wildemann B, et al. In contrast to effector T cells, CD4+CD25+FoxP3+ regulatory T cells are highly susceptible to CD95 ligand-but not to TCR-mediated cell death. J Immunol. 2005;175:32–6. doi: 10.4049/jimmunol.175.1.32. [DOI] [PubMed] [Google Scholar]
- 33.Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 34.Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, et al. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell. 2006;10:77–87. doi: 10.1016/j.ccr.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 35.Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101:1714–9. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khodarev NN, Minn AJ, Efimova EV, Darga TE, Labay E, Beckett M, et al. Signal transducer and activator of transcription 1 regulates both cytotoxic and prosurvival functions in tumor cells. Cancer Res. 2007;67:9214–20. doi: 10.1158/0008-5472.CAN-07-1019. [DOI] [PubMed] [Google Scholar]
- 37.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105:18490–5. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedberg JW, Dong DA, Li S, Kim H, Stephans K, Noonan K, et al. Oral fludarabine has significant activity in patients with previously untreated chronic lymphocytic leukemia, and leads to increased STAT1 levels in vivo. Leuk Res. 2004;28:139–47. doi: 10.1016/s0145-2126(03)00213-3. [DOI] [PubMed] [Google Scholar]
- 39.Thomas M, Finnegan CE, Rogers KM, Purcell JW, Trimble A, Johnston PG, et al. STAT1: a modulator of chemotherapy-induced apoptosis. Cancer Res. 2004;64:8357–64. doi: 10.1158/0008-5472.CAN-04-1864. [DOI] [PubMed] [Google Scholar]
- 40.Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, et al. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer. 2005;92:1149–58. doi: 10.1038/sj.bjc.6602447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fryknas M, Dhar S, Oberg F, Rickardson L, Rydaker M, Goransson H, et al. STAT1 signaling is associated with acquired crossresistance to doxorubicin and radiation in myeloma cell lines. Int J Cancer. 2007;120:189–95. doi: 10.1002/ijc.22291. [DOI] [PubMed] [Google Scholar]
- 42.Rickardson L, Fryknas M, Dhar S, Lovborg H, Gullbo J, Rydaker M, et al. Identification of molecular mechanisms for cellular drug resistance by combining drug activity and gene expression profiles. Br J Cancer. 2005;93:483–92. doi: 10.1038/sj.bjc.6602699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitroda SP, Wakim BT, Sood RF, Beveridge MG, Beckett MA, MacDermed DM, et al. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009;7:68. doi: 10.1186/1741-7015-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khodarev NN, Roach P, Pitroda SP, Golden DW, Bhayani M, Shao MY, et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS One. 2009;4:e5821. doi: 10.1371/journal.pone.0005821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Efimova EV, Liang H, Pitroda SP, Labay E, Darga TE, Levina V, et al. Radioresistance of Stat1 over-expressing tumour cells is associated with suppressed apoptotic response to cytotoxic agents and increased IL6-IL8 signalling. Int J Radiat Biol. 2009;85:421–31. doi: 10.1080/09553000902838566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owen-Schaub LB, van Golen KL, Hill LL, Price JE. Fas and Fas ligand interactions suppress melanoma lung metastasis. J Exp Med. 1998;188:1717–23. doi: 10.1084/jem.188.9.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caldwell SA, Ryan MH, McDuffie E, Abrams SI. The Fas/Fas ligand pathway is important for optimal tumor regression in a mouse model of CTL adoptive immunotherapy of experimental CMS4 lung metastases. J Immunol. 2003;171:2402–12. doi: 10.4049/jimmunol.171.5.2402. [DOI] [PubMed] [Google Scholar]
- 48.Seki N, Brooks AD, Carter CR, Back TC, Parsoneault EM, Smyth MJ, et al. Tumor-specific CTL kill murine renal cancer cells using both perforin and Fas ligand-mediated lysis in vitro, but cause tumor regression in vivo in the absence of perforin. J Immunol. 2002;168:3484–92. doi: 10.4049/jimmunol.168.7.3484. [DOI] [PubMed] [Google Scholar]
- 49.Fingleton B, Carter KJ, Matrisian LM. Loss of functional Fas ligand enhances intestinal tumorigenesis in the Min mouse model. Cancer Res. 2007;67:4800–6. doi: 10.1158/0008-5472.CAN-06-4473. [DOI] [PubMed] [Google Scholar]
- 50.Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A. 2009;106:9373–8. doi: 10.1073/pnas.0903487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.