

Abstract
We report the functional analysis of an artificial hexacoordinate oxygen transport protein, HP7, which operates via a mechanism similar to that of human neuroglobin and cytoglobin: the destabilization of one of two heme-ligating histidine residues. In the case of HP7 this is the result of the coupling of histidine side chain ligation with the burial of three charged glutamate residues on the same helix. Here we compare gaseous ligand binding, including rates, affinities and oxyferrous state lifetimes, of both heme binding sites in HP7. We find that despite the identical sequence of helices in both binding sites, there are differences in oxygen affinity and oxyferrous state lifetime which may be the result of differences in the freedom of motion imposed by the candelabra fold on the two sites of the protein. We further examine the effect of mutational removal of the buried glutamates on function. Heme iron in the ferrous state of this mutant is rapidly oxidized when when exposed to oxygen. Compared to HP7, distal histidine affinity is increased by a 22-fold decrease in the histidine ligand off-rate. EPR comparison of these ferric hemoproteins demonstrates that the mutation increases disorder at the heme binding site. NMR-detected deuterium exchange demonstrates that the mutation greatly increases water penetration into the protein core. The inability of the mutant protein to bind oxygen may be due to increased water penetration, the large decrease in binding rate caused by the increase in distal histidine affinity, or a combination of the two factors. Together these data underline the importance of the control of protein dynamics in the design of functional artificial proteins.
The first step in oxygen activation and/or transport by heme proteins is the binding of molecular oxygen to the ferrous heme iron while avoiding heme oxidation. It is critical to understand the underlying engineering parameters necessary for this process in order to design artificial oxygen-utilizing heme proteins. Moreover, many enzymes which utilize molecular oxygen as a substrate contain a number of redox-active cofactors in addition to the active site heme. For example the heme cofactors in cytochrome c oxidase serve to transport electrons into the O2-utilizing heme A-containing active site (1). Thus it will further be necessary in the future design of more complex artificial catalysts to be able to restrict gaseous ligand binding to those sites where catalysis is intended.
The family of hexacoordinate hemoglobins are oxygen-activating enzymes characterized by the property that they are bis-histidine-ligated in the oxidized state and exist in a mixed bis- and mono-histidine ligation state when reduced (2). The transient pentacoordination of the heme cofactor allows for the binding of molecular oxygen. We have recently reported the design, bacterial expression, and biochemical analysis of the completely artificial hexacoordinate oxygen transport protein HP7 (3). This protein consists of two heme cofactors bound to a homodimeric four alpha helix bundle protein. Each monomer is in a helix-loop-helix configuration and the two monomers bind to each other via hydrophobic sequestration with the monomer loops on the same end of the protein and attached to each other via a disulfide bond, a topology we have termed the ‘candelabra motif’ (4) (See Figure 1).
Figure 1.
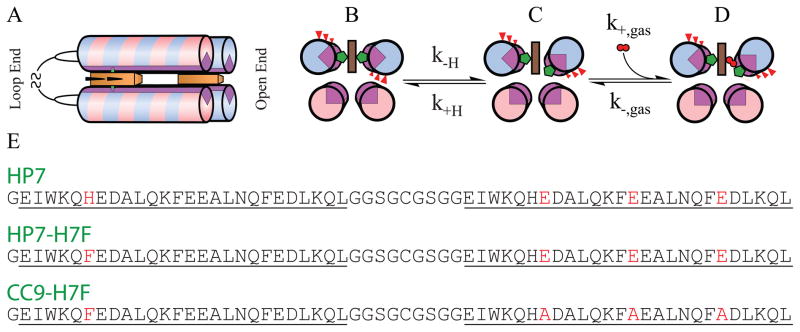
Structure, sequence and mechanism of HP7 and its mutants. (A) All three proteins are homodimers in a candelabra configuration in which the helix-loop-helix monomers are connected by a disulfide bond in the loop region. The open end binding site is defined as the end furthest from the loops, and the loop end binding is that closest. (B–D) Mechanism of gaseous ligand biding. State (B) is the entatic state. (E) Sequences of HP7, the distal heme binding site knockout mutant HP7-H7F, and the entatic state stabilizing mutant CC9-H7F. Helices are underlined and the b-position residues mutated in the transition from HP7 to CC9 are in red.
The heme cofactors bind to parallel helices via histidine ligand residues at the seventh position of each helix, one at the site furthest from the loops (the ‘open’ end) and one at the site closest to the loops (the ‘loop’ end).2 One of each pair of identical helices which bind a heme cofactor is oriented such that three polar glutamic acid residues must rotate into the hydrophobic core of the protein when the histidine is ligated to the heme iron. This strained, ‘entatic’ conformation (5) relaxes via the detachment of the distal histidine followed by rotation of the helix to move the glutamate side chains into solution, opening a ligation site on the heme iron to bind a gaseous ligand (Figure 1B–D).
The HP7 apoprotein binds two hemes sequentially, the first at the open end of the protein and the second near the loops (4). Our initial experiments were performed with a single heme bound to HP7 at the open end binding site. We then simplified the protein by removing the open end heme binding site, creating the homodimeric protein HP7-H7F which contains a single hexacoordinate binding site in the loop end of the bundle (6). Then, to examine the effects of changes in distal histidine ligand association energy on the overall heme binding affinity, we mutated the three buried glutamate residues on the ligating helix to alanine. This was found to slow the histidine ligand off-rate 22-fold, increase the affinity of the distal histidine ligand by a factor of thirteen and increase the bound cofactor reduction potential by 65 mV. However, it also decreased the heme binding affinity by a factor of five in the reduced state and 60 in the oxidized state. This was suggested to be caused by the homodimeric nature of the candelabra fold – mutations to the set of glutamates on the distal histidine helix responsible for the entatic state are also necessarily present in the proximal, nonrotating helix. The loss of these side chains greatly weakens binding, as evinced by a 60-fold decrease in the calculated ferrous heme binding constant to the histidine-detached pentacoordinate state, in which distal histidine ligation does not play a factor.
Here we compare gaseous ligand binding, including rates, affinities and oxyferrous state lifetimes, of both heme binding sites in HP7. While the intrinsic pentacoordinate affinity of each site for oxygen is identical, differences in the affinity of the distal histidine ligand cause the apparent affinities to differ by a factor of ten. The oxyferrous state lifetime is similar despite these differences, a result of an increased oxidation rate at the heme furthest from the connecting loops in the candelabra fold. We further show that by substituting the three strain-inducing glutamate residues we can effectively manipulate the thermodynamics of the entatic state and modulate the affinity and kinetics of the binding of carbon monoxide (CO), a nonreactive oxygen analogue. Oxygen binds to HP7 but not the mutant, although we cannot determine whether the inability of the mutant protein to bind O2 is a result of the greatly slowed rate of histidine ligand detachment or increased water penetration due to the larger degree of disorder caused by the mutation.
Materials and Method
Chemicals
Hemin was purchased from Fluka (Buchs, Switzerland). Molecular oxygen (O2) (99.98% purity), carbon monoxide (CO)1 (99.9%), and molecular nitrogen (N2) (99.99%) gases were from Matheson Gas (Basking Ridge, NJ) and the latter two were scrubbed of residual O2 by passage through two bubblers filled with a reduced vanadium sulfate solution followed by another filled with water (7). PD-10 desalting columns were from GE Healthcare (Port Washington, NY). All other solvents and reagents were from either Fisher Scientific or Sigma.
General biochemistry
The proteins HP7, HP7-H7F and CC9-H7F were expressed and purified and heme complexes were formed as described earlier (4, 6). Optical spectra were collected with a Hewlett-Packard (New York, NY) 8452A Diode array spectrophotometer running the Olis (Bogart, GA) SpectralWorks software and equipped with a Quantum Northwest (Liberty Lake, WA) Peltier temperature controller. All experiments were performed at 20° C in 250mM Boric Acid, 100mM KCl pH 9.0 unless otherwise specified. Each kinetic experiment was performed at least three times and reported errors are standard deviations from the mean.
Stopped-flow analysis of O2 binding
Binding kinetics of O2 with ferrous protein-heme complexes were followed spectroscopically in rapid stopped-flow mixing experiments over gas concentrations from 2% to 50% saturation at 15° C using a Biologic (Lyon, France) SFM 300 stopped flow mixer equipped with either a Biologic MOS 200 absorbance detector for single wavelength detection or a custom-built Olis RSM 1000 spectrometer for multiwavelength detection. Air- or gas-saturated buffer was mixed with degassed buffer in the first mix, and then this mixture was combined in a second mix with an anaerobic ferrous heme protein solution. Protein concentrations were 20 μM and ferrous complexes were prepared by carefully titrating anaerobic solutions of the holoproteins with a slight excess of sodium dithionite as observed by visible spectroscopy and then anaerobically transferred to the stopped-flow loading syringe by canula. Binding kinetic data were fit with Eqn. 1, which assumes that O2 binding rate, k+O2, is much greater than the sum of the distal histidine association and dissociation rates (2):
| (1) |
Where kobs is the fitted single exponential binding rate, k+H and k−H are the distal histidine-ferrous heme iron association and dissociation rates and k+O2 is the O2 association rate constant. At high [O2], kobs = k−H.
Kinetic analysis of O2 dissociation
The dissociation rate of the O2 complex was determined in rapid stopped-flow double mixing experiments using the CO displacement method (8). Briefly, the O2 complex was formed as above by mixing reduced protein and O2-saturated buffer. After a delay of 100 ms for complex formation this mixture was mixed with a two-fold larger volume of CO-containing buffer. The rate of O2 replacement by CO was collected using final CO concentrations ranging from 200–600 μM. At high CO concentration, the observed rate of replacement, robs is given by Eqn. 2:
| (2) |
When k+co[CO] ≫ k+O2[O2], the observed replacement rate constant is directly equal to O2 dissociation rate constant: robs≈ k−O2.
Oxyferrous state lifetimes
The lifetimes of O2-bound ferrous protein-heme complexes were followed spectroscopically in rapid stopped-flow mixing experiments using O2 concentrations from 16.67% to 66.67% saturation at 16° C using the stopped flow apparatus described above. Protein concentrations were 10 μM with 1 μM hemin added to ensure full complexation. Ferrous complexes were prepared by carefully titrating anaerobic solutions with a slight excess of sodium dithionite as observed by visible spectroscopy. Ferrous samples were anaerobically transferred to the stopped-flow loading syringe by canula. Kinetic data were fit with Eqn. 3, which is derived in the supplementary material:
| (3) |
Where [P] is the total protein concentration, [O2] is the total dissolved O2 concentration, Kd is the O2 dissociation constant, and kox is the second order rate constant for the oxidation of the protein-heme complex by O2 when not bound to O2.
Kinetic analysis of CO dissociation
The dissociation rate of the CO complex was determined using the ferricyanide trapping method of Moffet et al (9): briefly, anaerobic solutions of carbonmonoxyferrous protein complex were mixed with varying concentrations of potassium ferricyanide. The rates of the linked reactions of CO dissociation followed by heme oxidation by ferricyanide were followed by monitoring the disappearance of the carbonmonoxyferrous Soret peak at 421 nm. Double reciprocal plots of the oxidation rate vs. the concentration of ferricyanide extrapolated to infinite ferricyanide give the inverse CO dissociation rate as given by Eqn. 4:
| (4) |
Where kox,FC is the rate constant for the oxidation of CO-free complex by ferricyanide.
Flash photolysis analysis of CO association
The rate constants for both CO and histidine binding to the pentacoordinate state were determined using laser flash photolysis experiments performed as we’ve described previously (6): briefly a 1-ns pulse from a frequency doubled YAG laser excites the preformed carbonmonoxyferrous complex at 532 nm, causing the detachment of the ligand CO. This transiently forms an unliganded pentacoordinate heme protein and the rates of CO rebinding to the pentacoordinate state can be determined by analyzing the multi-exponential rebinding traces taken as a function of CO concentration using the method of Hargrove (10):
| (5) |
where γ1 and γ2 are the fitted first and second CO-dependent exponential rates and the kinetic constants are defined as in Equation 1 with the exception that CO denotes CO-binding rate constants instead of O2 binding rate constants. Protein concentrations were 20–25 μM and carbonmonoxyferrous complexes were prepared by titrating solutions of the holoproteins with an excess of dithionite as observed by visible spectroscopy under an atmosphere containing 10–100% CO mixed with argon. K+CO is taken directly from the slope of the replot of the sum of γ1 and γ2.
Electron paramagnetic resonance (EPR) spectroscopy
Low-temperature (7 K) EPR spectra were recorded on a Bruker E500 ElexSys EPR spectrometer operating at X-band using an Oxford Spectrostat continuous flow cryostat and ITC503 temperature controller. Data acquisition and manipulation were performed using XeprView and WinEPR software (Bruker). EPR samples were prepared by pipetting approximately 200 μL of 0.3mM protein solution in 200 mM TRIS-maleate buffer at pH 8.0 into 4 mm (3 mm i.d) precision bore quartz EPR tubes followed by immersion freezing in liquid nitrogen. Experimental parameters used were as follows: modulation amplitude, 4 G; microwave power, 1 mW; modulation frequency, 100 kHz; microwave frequency, 9.39 GHz; scan rate, 14.9 G/s; conversion time, 327 ms; time constant, 1310 ms. All spectra were obtained under identical instrumental conditions.
Hydrogen-Deuterium exchange
15N-labelled holo- and apoprotein samples of HP7-H7F and CC9-H7F in 25 mM potassium phosphate buffer pH 6.5 were passed through a PD-10 solvent exchange column pre-equilibrated in 25 mM KD2PO4 D2O buffer pD 6.5 (pH meter reading + 0.4 pH units), immediately placed in a 5mm NMR tube and equilibrated at 20°C for five minutes in the sample compartment of the NMR spectrometer. At this point protein concentrations were approximately 300 μM. The fraction of remaining amide protons was assessed as a function of time by collecting one dimensional HSQC, or isotope-selective, 1H spectra.
Results and Discussion
Protein design
HP7 complexed with one heme per homodimer has the open end binding site occupied (4). The HP7-H7F mutation eliminates the open end binding site but preserves the heme binding site nearest the loops. This enables the analysis and comparison of the the gaseous ligand binding properties of the two sites in the absence of complicating cooperative effects caused by interactions between the two hemes and the conformational effects, including helical rotation, caused by their binding. The CC9-H7F mutant has the three b-position glutamate residues on the histidine ligand helices of HP7-H7F mutated to alanine. This enables the analysis of gaseous ligand binding in the absence of a high-energy entatic state.
Comparison of the two heme sites in HP7
We have previously determined the on-rate, off-rate, and association constants for the distal histidine ligands of both sites in HP7 (see table 1) (3, 6). All four helices of HP7 have identical sequences and helical secondary structure as detected by NMR (4), which suggests that the functional characteristics of the two sites should be likewise identical. When a single heme is bound at the open end binding site, however, the distal histidine on-rate is two-fold faster and the off-rate is three-fold faster, resulting in a two-fold higher ligand association constant as compared to a single heme at the loop end site. We postulate that this might be a result of increased conformational freedom at the open end of the homodimer.
Table 1.
Haem iron ligand on- and off-rates and equilibrium constants at each site
Rates of O2 binding
We first compared the oxygen transport capability of the two different sites in HP7. Figure 2A depicts O2-dependence of the on-rate for molecular oxygen at each site. As equation 1 predicts, at higher O2 concentrations binding is rate-limited by the off-rate of the histidine ligand, and k+O2 is derived from the initial slope. O2 binds two-fold faster at high O2 concentrations and the pentacoordinate O2 binding rate is also faster when a single heme is bound at the open end of the protein, consistent with the more rapid distal histidine exchange rates at the open end which were observed earlier.
Figure 2.

Comparison of the on- and off-rates of molecular oxygen to both heme binding sites in HP7. (A) On-rates as a function of oxygen concentration. Reduced protein (20 μM), prepared by carefully titrating with a slight excess of dithionite, was mixed with oxygen in a stopped-flow and the rate of oxyferrous state formation followed spectroscopically. Lines shown are fits to the data with equation 1. (B) Off-rates as a function of carbon monoxide concentration. Lines drawn are the average rates of oxygen displacement.
Rates of O2 release
In our initial experiments (3), we measured the rate of CO replacement of O2 by cooling protein solutions containing 15% glycerol to low (−15 to 0° C) temperatures in a cuvette and then sequentially bubbling with O2 then CO. Rates of gaseous ligand exchange were followed spectroscopically as a function of temperature and the room temperature rate predicted by extrapolating to +16°C. Figure 2B depicts the results of double mixing experiments performed at +16°C which directly determine the off-rate of oxygen at this temperature. The reduced protein was first mixed with oxygen and then after a 100 ms delay for oxyferrous heme formation the complex was mixed with different concentrations of CO and the rate of CO complex formation followed spectroscopically. Because k+CO is much larger than k+O2, exchange rates are independent of the CO concentration over the range that experiments were performed, with the open end heme having an O2 off-rate more than three-fold higher than the loop end (see Figure 2B and Table 1). The rate we observe for O2 release from the open end heme is 12-fold faster than reported previously. This is likely due either to nonlinear behavior in the temperature range between 0° and 16°C or an effect of glycerol on the properties of the protein complex.
Furthermore, the off-rate is three-fold slower from the open end binding site. This is again consistent with increased flexibility at the open end, as the structural rearrangements involving binding, including possible hydrogen bonding between the bound oxygen and the detached distal histidine (11), should be eased in a less conformationally restrictive environment.
Oxygen affinity of the two sites
The affinities of oxygen binding to the pentacoordinate state at each site, calculated by taking the ratio of the experimentally determined on- and off-rates, are enumerated in Table 1. The pentacoordinate binding affinity is four-fold greater at the open site. The true binding constant for O2 to a hexacoordinate hemoglobin, which takes into account interference from the hexacoordinate state, is given by the relation (12):
| (6) |
As the distal histidine association constant is almost two-fold higher for the loop end binding site this results in an almost 10-fold increase in the affinity at the open end binding site.
Oxyferrous state lifetimes
A critical facet of oxygen transport protein function is the lifetime, once formed, of the oxyferrous state. When bound to oxygen the ferrous heme has a reduction potential too high to be oxidized by a second molecule of oxygen (13). After the oxygen dissociates, however, the ferrous heme is subject to oxidation in both the hexa- and pentacoordinate ligation states. Two parameters therefore affect the oxyferrous state lifetime: the oxygen dissociation constant and the oxidation rate of the ferrous heme when not bound to oxygen (see Eqn. 3). Another possible oxidation mechanism is the direct dissociation of superoxide (14). This mechanism results in an oxidation rate identical to that of equation 3, but the rate constant kox is a dissociation rate constant instead of an outer-sphere oxidation rate. Therefore the term kox encompasses rate constants from both mechanisms.
The rate of oxidation of the oxyferrous state collected as a function of the solution oxygen concentration at each heme binding site is depicted in Figure 3, and Table 2 depicts the values derived from this data. Fitted dissociation constants agree well with those determined using the ratio of on- and off-rates (Table 1). The open end heme site has an oxidation rate almost two times that of the loop end site. This again correlates with increased dynamics at the open end, as oxyferrous lifetimes are negatively affected by water penetration into the heme binding site, and proton donation to molecular oxygen lowers the energy of electron transfer (11). Despite this, the oxyferrous half-life is significantly longer due to the higher affinity of the O2 complex at the open end heme binding site.
Figure 3.

Rates of oxyferrous state breakdown at both heme binding sites in HP7. Reduced protein (20 μM), prepared by carefully titrating with a slight excess of dithionite, was mixed with oxygen in a stopped-flow and the rate of protein oxidation followed spectroscopically. Lines drawn are fits with equation 3. (A) Oxygen concentration dependence of the loop end site in HP7-H7F. (B) Oxygen concentration dependence of the open end site in HP7 with one heme bound.
Table 2.
Heme- histidine oxygen binding life time
| Protein | ligation | Rmax(s−1) | Kd,O2(mM) | kox(s−1mM−2) | t1/2(s)* |
|---|---|---|---|---|---|
| HP7 | Open end | 0.15 ± 0.01 | 0.037 ± 0.007 | 2300 ± 90 | 5.42 ± 0.04 |
| HP7H7F | Loop end | 0.19 ± 0.01 | 0.15 ± 0.03 | 1300 ± 50 | 4.71 ± 0.04 |
Oxyferrous state half-life in air (21% O2).
Effects of the triple glutamate placement
We have previously shown that replacent of the three b-position glutamate residues on the distal histidine-containing helix of HP7-H7F with alanine increases the affinity of the distal histidine ligand while slowing the histidine on- and off-rates (6). Upon mixing ferrous CC9-H7F with oxygen, either using a stopped-flow at temperatures from 2–20°C or in glycerol-containing solutions at −15°C, the protein rapidly oxidizes with no detectable oxyferrous state formation (not shown). This result is similar to what we observed in our earlier report where all six b-position glutamates were removed (3). The targeted nature of this more limited set of mutations, however, enables us to ascribe this loss of function to this subset of three glutamates.
We then set out to determine whether this failure to bind O2 in a stable complex is due to differences in gaseous ligand binding by examining the interaction between these two proteins and the nonreactive O2 analogue CO. While determining the distal histidine ligand off-rate, we previously showed that CO binds to both proteins, but 22-fold slower to CC9-H7F than HP7-H7F at high CO concentrations (see Table 3) (6).
Table 3.
CO association/dissociation rates and equilibrium constants
Rates of CO release
Figure 2B depicts the ferricyanide trapping analysis of CO release in the two proteins. Off-rates differ by a factor of three (Table 3), which is unsurprising as the CO-bound state already has the distal histidine detached, and is thus unaffected by the stability of the entatic state or the kinetics of histidine release. The slopes of the replots, which are indicative of the rates of electron transfer between the unbound state and ferricyanide (9), indicate that this oxidation is faster, perhaps as a result of the higher reduction potential of the heme bound to CC9-H7F.
CO-flash photolysis determination of k+co,pent
We have previously reported the rebinding kinetics, after laser flash photolysis, of the distal histidine in HP7-H7F and CC9-H7F (6). In CC9-H7F, HP7-H7F, and our original analysis of HP7 (3), a fast exponential process which is independent of the concentration of CO is observed. Similar behavior has been observed in mouse neuroglobin, and was ascribed to a relaxation process following a probable rearrangement caused by the change in heme iron planarity induced by detachment of the CO ligand (15, 16). In each case two slower exponential processes, each of which varies in rate and magnitude with CO concentration, were also observed. Figure 2A depicts the replots of the sum of these two processes for HP7-H7F and CC9-H7F. Eqn. 2 states that, k+co,pent, the binding rate of CO to the pentacoordinate state, can be derived from the slopes of these replots.
CO affinity
CO binding rates to the pentacoordinate complex of the two proteins differ only by 30%. This coupled with the CO release rate allows the calculation of the CO affinities for the pentacoordinate states of both proteins: HP7-H7F has a Kd of 300 nM and CC9-H7F has a Kd of 80 nM. As was true for O2 binding, the true binding constant for CO to hexacoordinate hemoglobins takes into account the hexacoordinate state (12):
| (7) |
The calculated dissociation constant for CO binding to CC9-H7F is three fold higher than in HP7-H7F (table 1): the twelve-fold larger histidine affinity is counterbalanced by the higher intrinsic CO affinity of the CC9-H7F heme, resulting in a small overall decrease in CO affinity.
As previously mentioned, CC9-H7F does not detectably form an oxyferrous complex. However, CC9-H7F is clearly capable of binding gaseous ligands, and the similarity in CO affinity of the two proteins suggests that O2 affinity may be similar as well, although CO is not capable of hydrogen bonding when bound to a ferrous iron (17) so a differential effect due to hydrogen bonding cannot be discounted. Given a similar binding affinity, Eqn. 3 predicts that a much higher oxidation rate is the cause of this inability to bind O2. Our results comparing the open and closed end of the protein suggest that protein dynamics and water penetration are the cause of the observed modest changes in oxidation rates. The large decrease in oxyferrous state lifetime in CC9-H7F suggests a similarly large increase in local dynamics. We thus set out to compare the dynamics of CC9-H7F with HP7-H7F spectroscopically.
Electron Paramagnetic Resonance
EPR of ferric heme proteins offers insight into the local ligand environment and geometry (4, 18). Figure 5 depicts the X-band EPR spectra of both ferric complexes. As expected based on the relatively small KA,his, both spectra contain a mixture of high- and low-spin Fe(III) complexes. The signal near g= 6 is like that of myoglobin and other 5-coordinate ferric heme examples containing an axially coordinated histidyl imidazole ligand. The rhombic signals with features in the range from g = 3.52 to 1.54 are typical of bis-histidine coordinated low-spin Fe(III) complexes.
Figure 5.

X-band EPR comparison of ferric HP7-H7F and CC9-H7F. As expected based on the relatively small KA,his, both spectra contain a mixture of high- and low-spin Fe(III) complexes. The signal near g= 6 is similar to that of myoglobin and other 5 ferric heme examples containing an axially coordinated histidyl imidazole ligand. The rhombic signals with features in the range from g = 3.52 to 1.54 are typical of bis-histidine coordinated low-spin Fe(III) complexes.
HP7-H7F exhibits two populations of low-spin hexacoordinate heme while CC9-H7F displays a much higher degree of conformational heterogeneity – there are at least three conformations of approximately equal population. Two populations are to be expected, as for a minimally designed protein such as these one would expect a mixture of two heme insertion isomers (19). The three different populations present in CC9-H7F, however, are indicative of a higher degree of disorder. It is not possible to tell by EPR alone whether this disorder is structural or dynamic in nature – the three different conformations might themselves be stable, with interchange between them occurring on such a slow time scale that water penetration is not accelerated. We therefore set out to spectroscopically compare the dynamic properties of these two proteins using NMR-detected hydrogen exchange.
Backbone hydrogen exchange
Figure 5 depicts NMR-detected amide proton hydrogen exchange data for both protein-ferrous heme complexes. Hydrogen exchange takes more than two hours to complete for the HP7-H7F complex while for CC9-H7F exchange is complete by the first time point in 30 minutes – a difference of at least a factor of ten in amide proton protection factor. As hydrogen exchange in the hydrophobic core of a protein is mainly limited by the frequency of its large-scale dynamics (20), this is an indication that the latter protein is also significantly more dynamic in the reduced state. Together, these data indicate that conformational heterogeneity leads to water penetration, which can lead to rapid oxidation of iron by proton donation and the failure to observe an oxyheme species.
Conclusions – Implications for protein design
These data demonstrate the beneficial and deleterious effects of dynamics on protein function in the same protein. This, we believe, demonstrates the importance of engineering dynamic motions into artificial proteins.
Flexibility and ligand binding
Gaseous ligand binding is faster and binding affinity is greater at the heme binding site near the open end of HP7, this despite the fact that the local sequence, secondary structure and cofactor reduction potentials are identical at both sites. We attribute this to increased flexibility at the less conformationally constrained open end binding site. Although the improvement demonstrated here is relatively modest, it underlines the important role dynamics play in ligand binding, and the need for protein flexibility to be incorporated into the protein design process.
Active site flexibility has been long been known to be an important factor in enabling rapid ligand binding in several natural enzymes (21) and in some limiting cases ligand binding even occurs to unfolded protein states, followed by folding (22). Several computationally designed proteins have been shown to be too rigid in their apo states to bind ligands on an experimentally tractable time scale: Bender et al, for example, were forced to heat their designed diporphyrin-binding four-helix bundle to 90°C for hours in the presence of denaturants in order to observe a significant level of active site occupancy (23), and McAllister et al heavily mutated their artificial tetraporphyrin-binding four-helix bundle in order to destabilize it enough to enable binding (24). In both cases dynamics had to be increased in order to achieve ligand binding, underlining the fact that what many protein design groups consider to be the optimal hydrophobic core packing – a relatively immobile semi-crystalline core with a minimum of both cavities and strained rotamers (25) – can lead to an inability to bind ligands on biologically relevant time scales.
In the case of HP7, the apoprotein is a molten globule which orders upon cofactor binding (4). This large degree of disorder explains the observed rapid heme binding. In the holo state, as the amino acid side chains at the active site differ significantly from those which bioinformatic analysis suggests would create a complementary binding interface (26), the relative ease of distal histidine association and dissociation and the ability of the pentacoordinate state to accommodate gaseous ligand binding can be explained by the relatively poor packing interactions which result from our simple binary patterning-based design process (18). It may indeed be an advantage of our design algorithm that hydrophobic core packing is not optimized. The observed improvement at the open end of the protein further supports this hypothesis.
Gaseous ligand binding in hexacoordinate hemoglobins
In hexacoordinate hemoglobins, gaseous ligand binding is affected both kinetically and thermodynamically by the association of the competing distal histidine. In terms of thermodynamics, equations 6 and 7 demonstrate that the effective oxygen binding affinity can be modulated via alterations in the distal histidine affinity. Given that the pentacoordinate oxygen affinities at the different sites of HP7 are almost identical, this may prove moving forward to be the simplest method to create artificial oxygen transport proteins with a range of oxygen dissociation constants.
While our data clearly demonstrate greatly increased dynamics and water penetration in the triple mutant protein, the kinetics of histidine dissociation can also play a role in function – the longer the dissociation takes the lower the oxyferrous state yield as the hexacoordinate state is vulnerable to oxidation. Thus an optimal hexacoordinate oxygen transport protein will have both the correct KA,HIS to achieve the target oxygen affinity, k−H must be fast enough to outcompete kox. One possible explanation for the inability of CC9-H7F to form an oxyferrous state is that the 22-fold increase in the average histidine detachment time gives too long a time for oxidation of the hexacoordinate complex prior to oxygen binding. However, we have also shown that increased disorder treatly accelerates water penetration into the hydrophobic core of the protein. We cannot determine which, or even both, of these mechanisms is serving to prevent buildup of a observable fraction of the oxyferrous state in CC9-H7F.
Supplementary Material
Figure 4.

Rates of CO complex formation and dissociation in HP7-H7F and CC9-H7F. (A) Replots of laser flash photolysis data from reference 6 used to calculate the pentacoordinate CO binding rate constant, k+CO. It is derived from the slope of the CO concentration dependence using equation 5. Open circles – CC9-H7F, closed triangles – HP7-H7F. (B) Ferricyanide trapping analysis of CO release. Double reciprocal plots of the rates of oxidation vs. the concentration of ferricyanide extrapolated to infinite ferricyanide give the CO dissociation rate as shown in equation 4 (9).
Figure 6.

Amide proton hydrogen exchange in HP7-H7F and CC9-H7F. Isotope-edited 1H-NMR spectra of 15N-labelled ferrous heme-bound HP7-H7F (top) and CC9-H7F (bottom) as a function of time after solvent exchange into 25 mM KaD2PO4 D2O buffer pD 6.5 at 20°C. Hydrogen exchange takes more than two hours to complete for the HP7-H7F complex while for CC9-H7F exchange is complete by the first time point in 30 minutes – a difference of at least a factor of ten in amide proton protection factor.
Acknowledgments
The authors thank Brian Gibney of the Department of Chemistry, Brooklyn College and Mark Hargrove, of the Department of Biophysics, Iowa State University for many helpful discussions. We thank Hsin Wang, of the Department of Chemistry, the City College of New York, for assistance with NMR measurements. We further thank an anonymous reviewer for several helpful comments.
Footnotes
Abbreviations used: CO, carbon monoxide; EPR, electron paramagnetic resonance; HSQC, heteronuclear single quantum coherence; NMR, nuclear magnetic resonance; O2, molecular oxygen.
We have chosen this terminology instead of proximal and distal to avoid confusion of the heme binding site location with the proximal and distal histidine ligand in each binding site.
RLK gratefully acknowledges support by the following grants: W81XWH-11-2-0083 from the Congressionally Directed Medical Research Program, infrastructure support from P41 GM-66354 to the New York Structural Biology Center and the National Institutes of Health National Center for Research Resources to the City College of New York (NIH 5G12 RR03060). EMEA gratefully acknowledges support from the City College of New York-Kungliga Tekniska Högskolan exchange program supported by the National Science Foundation Grant 0968244.
Supporting Information Available
Derivation of Equation 3, which describes the oxygen concentration dependence of the oxyferrous state lifetime. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Brunori M, Giuffre A, Sarti P. Cytochrome c oxidase, ligands and electrons. Journal Of Inorganic Biochemistry. 2005;99:324–336. doi: 10.1016/j.jinorgbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Trent JT, Hvitved AN, Hargrove MS. A model for ligand binding to hexacoordinate hemoglobins. Biochemistry. 2001;40:6155–6163. doi: 10.1021/bi0100790. [DOI] [PubMed] [Google Scholar]
- 3.Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, Dutton PL. Design and engineering of an O2 transport protein. Nature. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koder RL, Valentine KG, Cerda JF, Noy D, Smith KM, Wand AJ, Dutton PL. Native-like structure in designed four helix bundles driven by buried polar interactions. J Am Chem Soc. 2006;128:14450–14451. doi: 10.1021/ja064883r. [DOI] [PubMed] [Google Scholar]
- 5.Vallee BL, Williams RJP. Metalloenzymes - entatic nature of their active sites. Proc Natl Acad Sci U S A. 1968;59:498–505. doi: 10.1073/pnas.59.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Anderson JLR, Ahmed I, Norman JA, Negron C, Mutter AC, Dutton PL, Koder RL. Manipulating Cofactor Binding Thermodynamics in an Artificial Oxygen Transport Protein. Biochemistry. 2011;50:10254–10261. doi: 10.1021/bi201242a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Englander SW, Calhoun DB, Englander JJ. Biochemistry without oxygen. Anal Biochem. 1987;161:300–306. doi: 10.1016/0003-2697(87)90454-4. [DOI] [PubMed] [Google Scholar]
- 8.Gardner AM, Martin LA, Gardner PR, Dou Y, Olson JS. Steady-state and transient kinetics of Escherichia coli nitric-oxide dioxygenase (flavohemoglobin) - The B10 tyrosine hydroxyl is essential for dioxygen binding and catalysis. J Biol Chem. 2000;275:12581–12589. doi: 10.1074/jbc.275.17.12581. [DOI] [PubMed] [Google Scholar]
- 9.Moffet DA, Case MA, House JC, Vogel K, Williams RD, Spiro TG, McLendon GL, Hecht MH. Carbon Monoxide Binding by de Novo Heme Proteins Derived from Designed Combinatorial Libraries. Journal of the American Chemical Society. 2001;123:2109–2115. doi: 10.1021/ja0036007. [DOI] [PubMed] [Google Scholar]
- 10.Hargrove MS. A flash photolysis method to characterize hexacoordinate hemoglobin kinetics. Biophys J. 2000;79:2733–2738. doi: 10.1016/S0006-3495(00)76512-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson JLR, Koder RL, Moser CC, Dutton PL. Controlling complexity and water penetration in functional de novo protein design. Biochem Soc Trans. 2008;36:1106–1111. doi: 10.1042/BST0361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smagghe BJ, Halder P, Hargrove MS. Globins and Other Nitric Oxide-Reactive Proteins, Pt A. Elsevier Academic Press Inc; San Diego: 2008. Measurement of distal histidine coordination equilibrium and kinetics in hexacoordinate hemoglobins; pp. 359–378. [DOI] [PubMed] [Google Scholar]
- 13.McLendon G, Smith M. Outer-sphere electron transfer reactions of the isolated active-site heme octapeptide from cytochrome-C. Inorg Chem. 1982;21:847–850. [Google Scholar]
- 14.Eich RF, Li TS, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, Olson JS. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 15.Ascenzi P, Bocedi A, de Sanctis D, Pesce A, Bolognesi M, Marden MC, Dewilde S, Moens L, Hankeln T, Burmester T. Neuroglobin and cytoglobin - Two new entries in the hemoglobin superfamily. Biochemistry and Molecular Biology Education. 2004;32:305–313. doi: 10.1002/bmb.2004.494032050386. [DOI] [PubMed] [Google Scholar]
- 16.Du WH, Syvitski R, Dewilde S, Moens L, La Mar GN. Solution H-1 NMR characterization of equilibrium heme orientational disorder with functional consequences in mouse neuroglobin. J Am Chem Soc. 2003;125:8080–8081. doi: 10.1021/ja034584r. [DOI] [PubMed] [Google Scholar]
- 17.Collman JP, Fu L. Synthetic models for hemoglobin and myoglobin. Chemical Reviews. 1999;32:455–463. [Google Scholar]
- 18.Huang SS, Koder RL, Lewis M, Wand AJ, Dutton PL. The HP-1 maquette: From an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. Proc Natl Acad Sci U S A. 2004;101:5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamar GN, Budd DL, Viscio DB, Smith KM, Langry KC. Proton Nuclear Magnetic-Resonance Characterization of Heme Disorder in Hemoproteins. Proc Natl Acad Sci U S A. 1978;75:5755–5759. doi: 10.1073/pnas.75.12.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Englander SW. Protein folding intermediates and pathways studied by hydrogen exchange. Annu Rev Biophys Biomolec Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 21.Nowak T, Mildvan AS. NMR studies of selectively hindered internal motion of substrate analogs at the active site of pyruvate kinase. Biochemistry. 1972;11:2813–2819. doi: 10.1021/bi00765a013. [DOI] [PubMed] [Google Scholar]
- 22.Pozdnyakova I, Wittung-Stafshede P. Biological relevance of metal binding before protein folding. J Am Chem Soc. 2001;123:10135–10136. doi: 10.1021/ja016252x. [DOI] [PubMed] [Google Scholar]
- 23.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. De novo design of a single-chain diphenylporphyrin metalloprotein. J Am Chem Soc. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAllister KA, Zou HL, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, Therien MJ, Blasie JK, DeGrado WF. Using alpha-helical coiled-coils to design nanostructured metalloporphyrin arrays. J Am Chem Soc. 2008;130:11921–11927. doi: 10.1021/ja800697g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nanda V, Zahid S, Xu F, Levine D. Computational design of intermolecular stability and specificity in protein self-assembly. In: Johnson ML, Brand L, editors. Methods in Enzymology, Vol 487: Computer Methods, Pt C. 2011. pp. 575–593. [DOI] [PubMed] [Google Scholar]
- 26.Negron C, Fufezan C, Koder RL. Helical Templates for Porphyrin Binding in Designed Proteins. Proteins-Structure Function And Bioinformatics. 2009;74:400–416. doi: 10.1002/prot.22143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.