

Abstract
We demonstrate an approach to the assembly of DNA-containing polyelectrolyte multilayers (PEMs) that can be used to promote rapid release of DNA from surfaces. The approach is based on layer-by-layer incorporation of poly(acrylic acid) (PAA) to promote rapid film erosion in physiologically relevant media.
The layer-by-layer assembly of oppositely charged polymers on surfaces provides a powerful approach to the fabrication of functional thin films and surface coatings.1,2 These materials (called ‘polyelectrolyte multilayers’, or PEMs) are often stable in aqueous environments, and the robust nature of these assemblies has been exploited to design surfaces of interest in a broad range of fundamental and applied contexts.3–6 Past studies have also demonstrated, however, that the incorporation of functionality that promotes the destabilization of these assemblies can provide unique opportunities to design films that erode and/or promote localized and controlled release of incorporated agents.3–10 Research over the last decade has yielded a rich array of chemical approaches to the design of PEMs that erode or disintegrate upon exposure to a broad range of environmental stimuli.5–9 When combined, these approaches provide a robust and modular platform for the design of thin films and coatings that could be used to promote the localized delivery DNA, proteins, and other agents (e.g., from the surfaces of film-coated implants, etc.).5,6,9
We have demonstrated in past studies that it is possible to fabricate PEMs that erode and promote the release of plasmid DNA by incorporating layers of degradable polyamines (such as polymer 1) into PEMs.11–14 Our past studies demonstrate that (i) PEMs fabricated using DNA and polymer 1 (referred to hereafter as ‘polymer 1/DNA films’) erode and release DNA gradually (e.g., over ~2–4 days)11,13 and (ii) that surfaces coated with these films can be used to promote the surface-mediated transfection of cells in vitro.12,13 The hydrolyzable ester functionality in polymer 1 plays an important role in promoting film erosion,15 and we and others have reported that it is possible to both prolong and tune rates of erosion and release by changing the structures of the polymers used to fabricate these materials (e.g., by modifying polymer backbone16–19 or side chain14 structure). Using this approach, it is possible to design thin films (e.g., ~100 nm thick) that release DNA, proteins, or other agents over periods ranging from several days to several weeks or months.
Although gradual and sustained release of DNA is important in a broad range of contexts, approaches that promote rapid transfer of DNA from film-coated surfaces could also open the door to the design of films suited for use in applications that are time limited (e.g. short interventional procedures) or that otherwise require short delivery times (e.g., release over minutes or hours rather than days, weeks, or months). One approach to the design of PEMs that exhibit faster release profiles is to design new cationic polymers that promote film erosion more rapidly. A second (and potentially more general) approach would be to exploit the nature of the layer-by-layer assembly process to incorporate auxiliary agents (e.g., as ‘layers’) that can promote the disruption of ionic interactions more rapidly when these films are exposed to physiological pH and ionic strength. One advantage of this second approach is that agents that promote such ‘instability’ could potentially be used to expand the usefulness of existing cationic polymers (such as polymer 1) and tune accessible ranges of release profiles more broadly (e.g., by control over the amount of disrupting agent incorporated, as opposed to the design of a new cationic polymer for each desired release profile). Here, we demonstrate that the layer-by-layer incorporation of a weak poly(acid) into DNA-containing PEMs can be used to fabricate films that promote rapid release of DNA when incubated in physiologically relevant media.
The work reported here builds upon past studies demonstrating that polymer multilayers fabricated using weak poly(acid)s [e.g., poly(acrylic acid), PAA] can undergo substantial changes in structure upon changes in environmental pH.7,20–27 For films assembled through hydrogen-bonding interactions, changes in pH that result in ionization of PAA can disrupt hydrogen-bonding interactions and promote film disassembly.20–22 For ionically- crosslinked PEMs fabricated using PAA or copolymers containing weak poly(acids), increases or decreases in pH that lead to changes in ionization can be used to change the nature of ionic interactions and promote disassembly23–25 or other processes of film decomposition.25–27 Increases in the ionization of weak poly(acid)s in PEMs exposed to elevated pH have been used to deconstruct ‘sacrificial’ PEM layers to produce freestanding membranes;23 processes that lead to reduced ionization (e.g., at lower pH) can lead to film decomposition and transformations to porous morphologies in some films.26,27 On the basis of the diversity of behaviors observed in these past studies, we sought to determine the influence that incorporation of PAA could have on PEMs fabricated using polymer 1 and DNA.
To explore the feasibility of this approach, we performed a series of experiments to incorporate PAA into films fabricated using polymer 1 and a plasmid DNA construct encoding enhanced green fluorescent protein (EGFP). For these studies, we developed a ‘tetralayer’ approach to the layer-by-layer assembly of these multicomponent PEMs – that is, PAA was incorporated into the structures of these films by alternating the deposition of a ‘bilayer’ (or layer pair) of polymer 1 and PAA with the deposition of a ‘bilayer’ of polymer 1 and DNA (see ESI for additional details). This process was designed to yield films composed of repeating ‘tetralayers’, fabricated to have the general structure (1/PAA/1/DNA)x (where x is the number of tetralayers deposited). For these studies, films were fabricated using polymer solutions adjusted to values of pH ranging from 4.5 to 4.9 (see ESI for details) on planar silicon to permit characterization of film growth using ellispometry.
Fig 1 shows a plot of film thickness versus the number of polymer layers deposited for a film fabricated to have the structure (1/PAA/1/DNA)4 (open circles). Inspection of these data reveals film thickness to increase linearly to a final thickness of ~180 nm after the deposition of 16 polymer layers. For comparison, Fig 1 also shows the growth curve for films having the structure (1/DNA)8 (closed circles; fabricated in the absence of PAA). The thicknesses of these films (~100 nm) were generally consistent with results reported in past studies.11 The differences in the growth profiles of these two films suggest that PAA was incorporated into these materials during assembly.
Figure 1.

Plot of ellipsometric film thickness v. the number of layers of polymer deposited onto a silicon substrate for a (1/DNA)8 film (●) and a (1/PAA/1/DNA)4 film (○). Error bars represent the average of at least 5 different values measured over the length of a film. Silicon substrates were pre-coated with 10 bilayers of an LPEI/SPS (~ 30 nm thick) film prior to fabrication.
To characterize the DNA release profiles of these assemblies in physiologically relevant media, we incubated substrates coated with films having the structure (1/PAA/1/DNA)4 or (1/DNA)8 in phosphate-buffered saline (PBS, pH = 7.4) at 37 °C. Fig 2A shows a plot of the release of DNA into solution as a function of time. Inspection of these data reveals that (1/DNA)8 films released DNA over a period of ~2–3 days (closed circles), consistent with the release profiles reported in our past studies.11 In contrast, films having the structure (1/PAA/1/DNA)4 (open circles) released DNA much more rapidly over a period of ~3–6 h. Further inspection reveals these PAA-containing films to release approximately half the amount of DNA (~4 μg) as compared to polymer 1/DNA films (~8 μg). Although it is not possible to draw conclusions about the loading of DNA solely on the basis of initial film thickness, these differences are consistent with the fact that (1/PAA/1/DNA)4 films contain half the number of layers of DNA as compared to the (1/DNA)8 films. Fig 2B shows a plot of changes in film thickness versus time for these same film-coated substrates. These results reveal differences in the physical erosion profiles of these materials that correlate with the large differences in DNA release profiles shown in Fig 2A.
Figure 2.
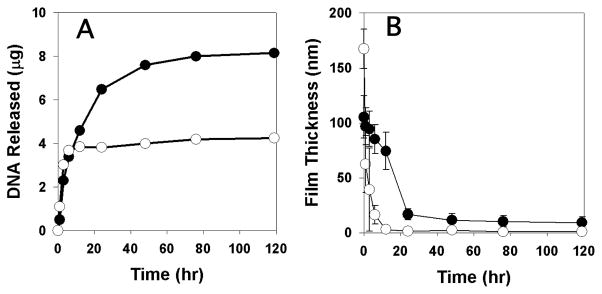
Plots of (A) release of plasmid DNA and (B) changes in film thickness for (1/DNA)8 films (●) and (1/PAA/1/DNA)4 films (○) incubated in PBS at 37 °C. Error bars represent deviations in film thickness at five positions along the length of a film.
The results above, when combined, support the view that PAA is incorporated into these PEMs during layer-by-layer assembly. Our results also demonstrate that incorporation of PAA results in DNA-containing films that erode or disintegrate rapidly when incubated in PBS. As discussed above, films having the structure (1/DNA)8 generally erode and release DNA over a period of ~2 days11 (a process that is understood to be promoted, at least in part, by the time-dependent hydrolysis of the ester bonds in the backbone of polymer 1).15,28 The erosion and release of DNA from our PAA-containing films, however, is sufficiently rapid that film erosion is less likely to depend on polymer hydrolysis.
The percent ionization of PAA generally increases as pH is increased (deprotonation leads to a more negatively charged polymer) and ionization is reduced at lower pH. Because the films used here were fabricated using solutions adjusted to values of pH (e.g., 4.5–4.9) that were substantially lower than that used in the release studies above (pH = 7.4), our results are consistent with the possibility that increased ionization of PAA at higher pH could lead to an increase in anionic charge in the films and change the nature of interactions in these ionically-crosslinked materials.23,25 The compositions and structures of our films differ considerably from those used in past studies on the pH-induced deconstruction of PEMs fabricated using weak poly(acid)s.23,25 We speculate, however, that net increases in uncompensated anionic charge resulting from ionization of PAA at elevated pH are sufficient to introduce repulsive interactions that lead to more rapid film erosion (relative to films that do not contain PAA). We caution, however, that our results do not rule out other possibilities (e.g., increases in internal osmotic pressure, rapid loss of PAA, or other processes resulting from increased ionization)25 and that, in general, PEMs assembled from weak polyelectrolytes represent exceptionally complex systems. For example, while past studies report the pKa of PAA in dilute solution to be ~6.5,29 the degree of ionization of weak polyelectrolytes can sometimes change substantially upon incorporation into a PEM.25,29 In addition, we note that while past studies of PAA-containing multilayers have used FTIR spectroscopy to characterize changes in the ionization of PAA,20,23,29 the additional complexity of our films (e.g., multiple carbonyls arising from DNA, polymer 1, and PAA) complicates characterization using these methods. Additional studies will be required to characterize changes in ionic interactions in these films more completely. In the context of this study, however, we do conclude that PAA can be incorporated into polymer 1/DNA films in a manner that permits rapid disassembly under conditions relevant in the context of potential biomedical applications.
Fig 3A shows a representative fluorescence micrograph of mammalian cells treated with a sample of DNA released from a (1/PAA/1/DNA)4 film 3 h after incubation in PBS (see ESI for additional details). The green cells in this image correspond to cells expressing EGFP, and demonstrate that DNA is released from these PAA-containing films in a form that is able to promote transgene expression. These results suggest opportunities to use these rapidly-eroding films for the localized or contact-mediated transfer of DNA to cells12 (e.g., by placing film-coated substrates in contact with cells in vitro or in vivo).
Figure 3.
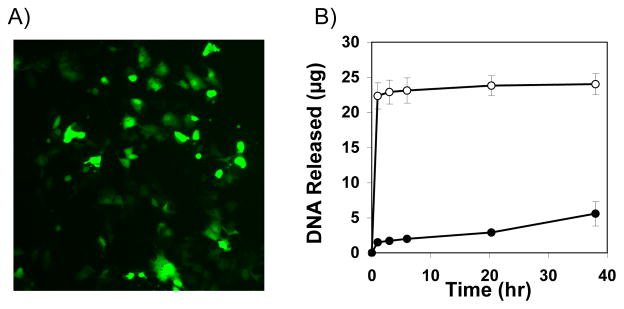
(A) Fluorescence micrograph (10X) of COS-7 cells (>95% confluent) treated with DNA released from a (1/PAA/1/DNA)4 film incubated in PBS for 3 h (see text and Supp. Info. for details). (B) Plot showing the release of DNA for (LPEI/DNA)8 films (●) and (LPEI/PAA/LPEI/DNA)4 films (○) incubated in PBS at 37 °C.
As discussed above, films fabricated using polymer 1 and DNA do erode and release DNA gradually, even in the absence of PAA. We conducted a final series of experiments to investigate the generality of this PAA-based approach and determine whether it could also be used to promote the disruption of DNA- containing PEMs that are otherwise stable in PBS (e.g., films fabricated using cationic polymers that are not degradable or do not incorporate other mechanisms to promote film erosion). For these studies, we selected films fabricated using DNA and linear poly(ethylene imine) (LPEI). We selected LPEI for two reasons: (i) LPEI is a polyamine used widely for the assembly of PEMs,2 and (ii) LPEI is used broadly for the delivery of DNA to cells. In this latter context, the design of PEMs that release DNA and well-known delivery agents could prove useful for the design of surfaces that promote more effective surface-mediated delivery.
To evaluate this approach, we fabricated films having the structure (LPEI/DNA)8 and (LPEI/PAA/LPEI/DNA)4 on silicon substrates. Fig 3B shows a comparison of the DNA-release profiles of these films upon incubation in PBS and reveals striking differences in behavior. While PAA-containing films released DNA rapidly and completely within the first 3 h of incubation, (LPEI/DNA)8 films did not release significant amounts of DNA for up to ~40 h (see Fig S1 of the ESI for additional physical erosion profiles). These results demonstrate that incorporation of PAA can also be used to promote rapid release of DNA from films fabricated using non-degradable cationic polymers that are otherwise significantly more stable.
We note here that while the results of this study do demonstrate that the incorporation of PAA can be used to promote rapid disassembly of DNA-containing PEMs, they do not provide direct insight into (i) the location of PAA in the films or (ii) the nature of the aggregates that might be released (e.g., whether DNA is released in a form that remains bound to polymer 1 or LPEI). Both of these issues will be important with respect to optimizing this approach and designing films for the surface-mediated delivery of DNA. In this context, we also note that our ‘tetralayer’ approach to the incorporation of weak poly(acid)s creates additional opportunities for the incorporation of other agents that could help promote intracellular processing of DNA. For example, poly(propylacrylic acid) (PPAA), a more hydrophobic analog of PAA, has been demonstrated to have pH-dependent membrane disrupting properties30,31 useful for enhancing intracellular delivery of a range of agents. Incorporation of PPAA or other membrane-active poly(acid)s into the structures of PEMs could thus contribute to the development of these materials for delivery-oriented applications.
Finally, we note again that a broad range of approaches have been developed to disrupt ionic interactions and impart ‘instability’ to PEMs under a variety of different conditions (e.g., actively, passively, or in response to specific stimuli).5–9 Approaches that can be used to trigger rapid release of DNA at physiological pH and ionic strength (without the need for external triggers) could be useful for the surface-mediated delivery of DNA in applications that require short delivery times (e.g., surgical procedures limited to short-term placement and removal of interventional devices) or in a range of other contexts. Studies to characterize the structures of these PAA-containing assemblies and investigate the ability of these films to promote localized and surface-mediated cell transfection are currently underway.
Supplementary Material
Acknowledgments
Support was provided by the NIH (R01 EB006820), the Alfred P. Sloan Foundation, and the UW Vilas Trust.
Footnotes
Electronic Supplementary Information (ESI) available: Full details of experimental protocols and additional characterization experiments. See DOI: 10.1039/b000000x/
Notes and references
- 1.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 2.Bertrand P, Jonas A, Laschewsky A, Legras R. Macromol Rapid Commun. 2000;21:319–348. [Google Scholar]
- 3.Ai H, Jones S, Lvov Y. Cell Biochemistry and Biophysics. 2003;39:23–43. doi: 10.1385/CBB:39:1:23. [DOI] [PubMed] [Google Scholar]
- 4.Peyratout CS, Dähne L. Angew Chem Intl Ed. 2004;43:3762–3783. doi: 10.1002/anie.200300568. [DOI] [PubMed] [Google Scholar]
- 5.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203–3224. [Google Scholar]
- 6.Boudou T, Crouzier T, Ren KF, Blin G, Picart C. Adv Mater. 2010;22:441–467. doi: 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- 7.Sukhishvili SA. Curr Opin Colloid Interface Sci. 2005;10:37–44. [Google Scholar]
- 8.De Geest BG, Sanders NN, Sukhorukov GB, Demeester J, De Smedt SC. Chem Soc Rev. 2007;36:636–649. doi: 10.1039/b600460c. [DOI] [PubMed] [Google Scholar]
- 9.Lynn DM. Adv Mater. 2007;19:4118–4130. [Google Scholar]
- 10.Jewell CM, Lynn DM. Adv Drug Delivery Rev. 2008;60:979–999. doi: 10.1016/j.addr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 12.Jewell CM, Zhang J, Fredin NJ, Lynn DM. J Controlled Release. 2005;106:214–223. doi: 10.1016/j.jconrel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 13.Jewell CM, Zhang J, Fredin NJ, Wolff MR, Hacker TA, Lynn DM. Biomacromolecules. 2006;7:2483–2491. doi: 10.1021/bm0604808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Montañez SI, Jewell CM, Lynn DM. Langmuir. 2007;23:11139–11146. doi: 10.1021/la702021s. [DOI] [PubMed] [Google Scholar]
- 15.Zhang JT, Fredin NJ, Lynn DM. J Polym Sci Pol Chem. 2006;44:5161–5173. [Google Scholar]
- 16.Zhang J, Fredin NJ, Janz JF, Sun B, Lynn DM. Langmuir. 2006;22:239–245. doi: 10.1021/la052360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Lynn DM. Macromolecules. 2006;39:8928–8935. doi: 10.1021/ma061815g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chuang HF, Smith RC, Hammond PT. Biomacromolecules. 2008;9:1660–1668. doi: 10.1021/bm800185h. [DOI] [PubMed] [Google Scholar]
- 19.Macdonald M, Rodriguez NM, Smith R, Hammond PT. J Controlled Release. 2008;131:228–234. doi: 10.1016/j.jconrel.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sukhishvili SA, Granick S. J Am Chem Soc. 2000;122:9550–9551. [Google Scholar]
- 21.Cho J, Caruso F. Macromolecules. 2003;36:2845–2851. [Google Scholar]
- 22.Ono SS, Decher G. Nano Lett. 2006;6:592–598. doi: 10.1021/nl0515504. [DOI] [PubMed] [Google Scholar]
- 23.Dubas ST, Farhat TR, Schlenoff JB. J Am Chem Soc. 2001;123:5368–5369. doi: 10.1021/ja015774+. [DOI] [PubMed] [Google Scholar]
- 24.Dubas ST, Schlenoff JB. Macromolecules. 2001;34:3736–3740. [Google Scholar]
- 25.Sui ZJ, Schlenoff JB. Langmuir. 2004;20:6026–6031. doi: 10.1021/la0495985. [DOI] [PubMed] [Google Scholar]
- 26.Mendelsohn JD, Barrett CJ, Chan VV, Pal AJ, Mayes AM, Rubner MF. Langmuir. 2000;16:5017–5023. [Google Scholar]
- 27.Hiller JA, Mendelsohn JD, Rubner MF. Nat Mater. 2002;1:59–63. doi: 10.1038/nmat719. [DOI] [PubMed] [Google Scholar]
- 28.Fredin NJ, Zhang J, Lynn DM. Langmuir. 2007;23:2273–2276. doi: 10.1021/la0624182. [DOI] [PubMed] [Google Scholar]
- 29.Choi J, Rubner MF. Macromolecules. 2005;38:116–124. [Google Scholar]
- 30.Seki K, Tirrell DA. Macromolecules. 1984;17:1692–1698. [Google Scholar]
- 31.Murthy N, Robichaud JR, Tirrell DA, Stayton PS, Hoffman AS. J Controlled Release. 1999;61:137–143. doi: 10.1016/s0168-3659(99)00114-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.