

Abstract
Ovarian cancer is the most lethal gynecological malignancy, characterized by a high rate of chemoresistance. Current treatment strategies for ovarian cancer focus on novel drug combinations of cytotoxic agents and molecular targeted agents or novel drug delivery strategies that often involve intraperitoneal (IP) injection. Poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) micelles were loaded with paclitaxel (cytotoxic agent), cyclopamine (hedgehog inhibitor), and gossypol (Bcl-2 inhibitor). After physicochemical studies focusing on combination drug solubilization, 3-drug PEG-b-PCL micelles were evaluated in vitro in 2-D and 3-D cell culture and in vivo in xenograft models of ovarian cancer, tracking bioluminescence signals from ES-2 and SKOV3 human ovarian cancer cell lines after IP injection. 3-drug PEG-b-PCL micelles were not significantly more potent in 2-D cell culture in comparison to paclitaxel; however, they disaggregated ES-2 tumor spheroids, whereas single drugs or 2-drug combinations only slowed growth of ES-2 tumor spheroids or had no noticeable effects. In ES-2 and SKOV3 xenograft models, 3-drug PEG-b-PCL micelles had significantly less tumor burden than paclitaxel based on bioluminescence imaging, 3′-deoxy-3′-18F-fluorothymidine (18F-FLT) PET imaging, and overall survival. 18F-FLT-PET images clearly showed that 3-drug PEG-b-PCL micelles dramatically reduce tumor volumes over paclitaxel and vehicle controls. In summary, PEG-b-PCL micelles enable the IP combination drug delivery of paclitaxel, cyclopamine and gossypol, resulting in tumor growth inhibition and prolonged survival over paclitaxel alone. These results validate a novel treatment strategy for ovarian cancer based on drug combinations of cytotoxic agents and molecular targeted agents, delivered concurrently by a nanoscale drug delivery system, e.g. PEG-b-PCL micelles.
Keywords: Bioluminescence imaging, combination drug delivery, ovarian cancer, paclitaxel, PET imaging, polymeric micelles
1. Introduction
Ovarian cancer is the most lethal female gynecological malignancy. The estimated death of ovarian cancer patients in 2011 was 6% based on US mortality data. From 1999 to 2006, localized, regional, and distant ovarian cancers were diagnosed at 15%, 17%, and 62%, respectively, in the United States, and the overall 5-year survival rates for ovarian cancer stand at 94% for localized, 73% for regional, and 28% for distant ovarian cancers [1]. Most ovarian tumors initially respond to conventional cytotoxic agents, such as taxane and platinum analogues. However, ovarian cancer that relapses after first-line treatment presents reduced response to cytotoxic agents and eventually recurs as a more aggressive disease [2].
Current treatment strategies for ovarian cancer in preclinical and clinical studies involve novel drug combinations that seek to overcome drug resistance of cytotoxic agents by targeting cancer cell survival signaling pathways with molecular targeted agents. For example, the hedgehog (Hh) pathway is activated in several malignancies and cancer stem cells, and it has recently been shown that the inhibition of Hh signaling results in reduced growth of ovarian cancer spheroid-forming cells: 10-, 5-, and 4-fold reduction in growth for ES-2, SKOV3, and TOV112D cells, respectively [3]. Inhibition of Hh signaling in ovarian cancer has recently been shown to reverse taxane resistance in several cell lines by what appears to be a reduction in p-glycoprotein expression (MDR1) [4]. In another cancer cell survival pathway, overexpression of anti-apoptotic Bcl-2 proteins is a common hallmark of many malignancies, and acquired cisplatin resistance in ovarian cancer cells has been linked to Bcl-2 overexpression [5]. Not surprisingly, interest in drug combinations of targeted agents with different mechanisms of action along with cytotoxic agents has developed, given tumor heterogeneity and redundancy in cancer cell signaling, with the caveat that dose-limiting toxicity is not overlapping.
At the same time, progress has been made in drug delivery strategies for the treatment of ovarian cancer, relying mostly on the intraperitoneal (IP) route [6]. Metastasis of ovarian cancer proceeds primarily via the peritoneal cavity, as opposed to the vascular system for other malignancies [7]. Thus, IP injection or infusion of cytotoxic agents enables direct access to ovarian cancer, reduced systemic exposure, and increased overall survival relative to intravenous injection of cytotoxic agents. More recently, De Souza et al. have shown in xenograft models that an IP, sustained release or continuous exposure of docetaxel reduces tumor mRNA-expression of genes encoding drug efflux transporters, e.g. MDR1 [8]. Lastly, Au and co-workers have championed the strategy of “tumor priming” for bulky peritoneal tumors and shown that apoptosis induction by paclitaxel (PTX) may promote tumor penetration of drugs, drug-containing nanoparticles, and drug-containing microparticles, overcoming barriers to transport in tumor interstitium and drug resistance owing to inadequate drug delivery [9, 10].
In this work, we show that poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) micelles have a carrying capacity for PTX, cyclopamine (CYP), and gossypol (GSP) (Fig. 1). CYP antagonizes the Hh pathway by directly binding to Smoothened in a Patched-independent manner [11]. GSP is a polyphenolic pro-apoptotic compound, inhibiting anti-apoptotic proteins (Bcl-2, Bcl-xL, and Mcl-1) [12, 13]. All three anticancer agents are poorly water-soluble, and PEG-b-PCL micelles can deliver all three together following multiple drug solubilization, enabling in vivo toxicity and efficacy studies in xenograft models. Our results corroborate previous studies on 3-drug poly(ethylene glycol)-block-poly(d,l-lactic acid) micelles, which also had a capacity for multiple drug solubilization. Tail vein injection of 3-drug poly(ethylene glycol)-block-poly(d,l-lactic acid) micelles resulted in robust antitumor responses in breast and lung cancer xenograft models [14]. In this work, we evaluated for the first time antitumor responses after IP injection of PEG-b-PCL micelles containing PTX, CYP, and GSP in ES-2 and SKOV3 xenograft models, assessing tumor burden by noninvasive optical bioluminescence imaging, micro-positron emission tomography/computed tomography (microPET/CT) imaging, and overall survival. Our results suggest that IP delivery of polymeric micelles carrying multiple anticancer agents is a highly promising treatment strategy for ovarian cancer, benefiting from novel drug combinations that act on drug resistance pathways and also regional and sustained drug release for enhanced intratumoral drug delivery.
Fig 1.
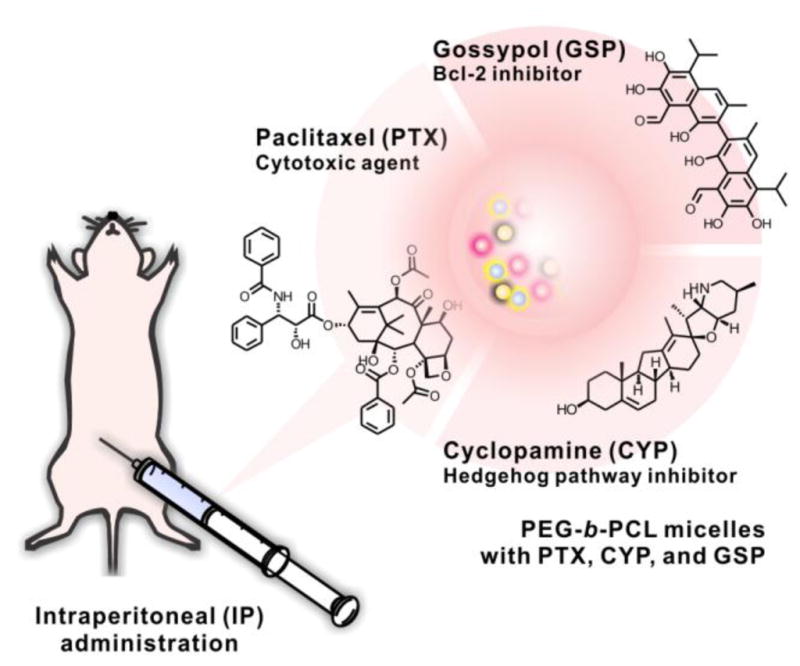
3-drug PEG-b-PCL micelles with PTX, CYP, and GSP, injected IP in ovarian xenograft models.
2. Materials and methods
2.1. Materials
PTX, CYP, and GSP were purchased from LC Laboratories (Woburn, MA), Logan Natural Products (Plano, TX), and LKT Laboratories, Inc (St. Paul, MN), respectively. PEG-b-PCL (Mn of PEG = 5,000 g/mol; Mn of PCL = 10,000 g/mol; and Mw/Mn = 1.3) was purchased from Polymer Source (Dorval, Canada). Dialysis cassettes (MWCO 20,000) were purchased from Thermo Fisher Scientific Inc (Rockford, IL). (ES-2 and SKOV3 human ovarian cancer cells were purchased from ATCC (Manassas, VA). Luciferase-expressing plasmid pGL4.51 containing the neomycin-resistance gene and luciferase assay system kit were purchased from Promega (Madison, WI). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). D-luciferin was purchased from Caliper Life Science (Hopkinton, MA). 3′-deoxy-3′-18F-fluorothymidine (18F-FLT) was purchased from the University of Wisconsin Cyclotron Research Center (Madison, WI). All other reagents were obtained from Thermo Fisher Scientific Inc. (Fairlawn, NJ) and were of analytical grade.
2.2. Methods
2.2.1. Preparation and characterization of drug-loaded PEG-b-PCL micelles
Drug-loaded PEG-b-PCL micelles were prepared by a solvent evaporation technique as previously described [15]. Briefly, 6.0 mg of PTX, CYP, and GSP each and 180 mg of PEG-b-PCL were dissolved in 1.0 mL of acetone, followed by a rapid addition of 1.0 mL of pre-warmed 0.9% saline or PBS (10 mM, pH 7.4) at 60 °C with vigorous mixing. Acetone was evaporated under reduced pressure using a rotatory evaporator at 60 °C. Insoluble drugs were removed by centrifugation for 5 min at 10,000 × g, followed by filtration with 0.22 μm nylon syringe filters. The content of PTX, CYP, and GSP in PEG-b-PCL micelles was quantified by reverse-phase HPLC (RP-HPLC) analysis with a Shimadzu Prominence HPLC system (Shimadzu, Japan). Samples (10 μL) were injected into a Zorbax SB-C8 rapid resolution cartridge (4.6 mm × 75 mm, 3.5 μm, Agilent). The flow rate was 1.0 mL/min, and the column was kept at 40 °C. The separation of PTX, CYP, and GSP was done in an isocratic mode with mobile phase consisting of 55% of acetonitrile, 45% distilled water, and 0.1% trifluoroacetic acid. PTX, CYP, and GSP were monitored at 227, 204, and 373 nm, respectively, and eluted at 2.7 min, 1.9 min, and 10.6 min, respectively.
Z-average diameters of PEG-b-PCL micelles at 25 °C were determined by dynamic light scattering (DLS) measurements using a Zetasizer Nano-ZS (Malvern Instruments, UK) at a fixed angle of 173°. Autocorrelation functions were treated by cumulant analysis, calculating the hydrodynamic diameter of PEG-b-PCL micelles from the Stokes-Einstein equation and the polydispersity index (PDI). Prior to measurements, PEG-b-PCL micelle solutions were diluted 10 times with 0.9% saline or PBS (10 mM, pH 7.4), resulting in the level of PEG-b-PCL at 1.8 mg/mL (above the critical micelle concentration).
The physical stability and drug release kinetics of drug-loaded PEG-b-PCL micelles were studied in vitro. An aqueous solution of drug-loaded PEG-b-PCL micelles (6.0 mg/mL of each drug) was incubated at 25 °C for 24 h and centrifuged for 5 min at 10,000 × g, followed by filtration using 0.22 μm nylon syringe filters to remove any precipitated drug. The level of drug remaining in solution after 24 h was quantified by RP-HPLC analysis as described above, and the % remaining drug in solution was calculated. In in vitro drug release experiments, an aqueous solution of drug-loaded PEG-b-PCL micelles (0.6 mg/mL of each drug) was put into dialysis cassettes (MWCO 20,000), and cassettes were placed in 2.0 L of PBS (10 mM, pH 7.4) at 37 °C with stirring. Samples (20 μL) were withdrawn, and cassettes were replenished with 20 μL of fresh PBS. Withdrawn samples at various time points, 0, 0.5, 1, 2, 3, 6, 15, 18, 24, 48, and 72 h, were analyzed by RP-HPLC to quantify the level of drug left in the dialysis cassette. Assuming that drug release from PEG-b-PCL micelles was rate-limiting, % drug release was calculated over time, and curve-fitting and estimation of time for 50% drug release (t1/2) were done based on a two-phase exponential association model using GraphPad Prism version 5.00 for Mac OS X (San Diego, CA).
2.2.2. Luciferase-expressing ES-2 and SKOV3 cells (ES-2-luc and SKOV3-luc)
ES-2 and SKOV3 human ovarian cancer cell lines were cultured in McCoy’s 5a medium and RPMI-1640 medium, respectively, supplemented with 1% L-glutamine, 10% fetal bovine serum, and 1% penicillin/streptomycin. ES-2 and SKOV3 cells were maintained at 37 °C under an atmosphere of 5% CO2 in a humidified incubator. Both ovarian cancer cell lines were stably transfected with luciferase-expressing plasmid pGL4.51 containing the neomycin-resistance gene (Promega, Madison, WI) using lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. After 48 h, ES-2 or SKOV3 cells were cultured in corresponding medium containing 750 μg/mL G418 selective antibiotics for 3 to 4 weeks until cell foci were obtained. Several G418-resistant clones were isolated and maintained in G418-containing medium at 500 μg/mL. Luciferase expression for expanded clones was quantified using a luciferase assay system kit (Promega, Madison, WI). The clone with the highest luciferase expression for each cell line was chosen for subsequent in vitro and in vivo experiments.
2.2.3. In vitro cytotoxicity study
In vitro cytotoxicity of drug-loaded PEG-b-PCL micelles was assessed in 2-D cell culture with luciferase-expressing ES-2-luc and SKOV3-luc cells and with 3-D tumor spheroids formed from ES-2-luc cells. SKOV-3-luc (10,000 cells/well) and ES-2-luc (5,000 cells/well) were seeded in 96-well plates and incubated for 24 h to form 2-D monolayers. ES-2-luc cells (1,000 cells/well) were suspended in agarose-coated 96-well plates and incubated for 4 days to generate 3-D tumor spheroids [16]. Drug-loaded PEG-b-PCL micelles were added at final drug concentrations of 0.1, 1, 10, 100, and 1000 nM, and cell viability was determined 72 h post drug treatment by quantifying the intensity of bioluminescence signal expressed in surviving cells using Xenogen IVIS® 200 Series (Caliper Life Science, Hopkinton, MA). D-luciferin at 15 μg/well was added to 96-well plates 5 min prior to bioluminescence imaging. The half maximal inhibitory drug concentration (IC50) was determined with the median effect equation provided by GraphPad Prism. The morphology of ES-2-luc tumor spheroids 3 days post drug treatment was observed by an inverted light microscope (Nikon, Japan).
2.2.4. Human ovarian cancer xenografts and drug treatment
Female 6–8 week-old athymic nude-Foxn1nu mice were purchased from Harlan Laboratories (Madison, WI). General anesthesia was induced with 1.5% isoflurane/oxygen and maintained with 1% isoflurane/oxygen. All animal experiments were approved by UW-Madison’s Institutional Animal Care and Use Committee and conducted in accordance with institutional and NIH guidance. ES-2-luc and SKOV3-luc cells were harvested from sub-confluent cultures after trypsinization, and ES-2-luc (1 × 106 cells/animal) and SKOV3-luc (2 × 106 cells/animal) cells were injected (approximately 100 μL) IP into anesthetized mice. Drug treatment was initiated 4 days and 16 days post cell inoculation of ES-2-luc cells and SK-OV-3-luc cells, respectively, after observation of bioluminescence in whole-body images of animals by Xenogen IVIS® 200 Series. Both ES-2-luc-bearing and SKOV-3-luc-bearing xenografts were divided into 3 groups (n = 4): 3-drug PEG-b-PCL micelles with PTX, CYP, and GSP at 30, 30, and 30 mg/kg; PEG-b-PCL micelles with paclitaxel at 30 mg/kg; and empty PEG-b-PCL micelles as a control. Each (approximately 200 μL) was injected IP into anesthetized nude mice q7d × 3 (days 0, 7, and 14). Body weights and radii of abdomen of mice were recorded up to 2 months by a portable scale and a digital caliper (Fisher Scientific, Pittsburgh, PA), respectively. All mice used for treatment response evaluations were euthanized at the time of reaching a moribund condition, and Kaplan-Meier survival curves were constructed.
2.2.5. Whole-body bioluminescence imaging
Animals were anesthetized as described above. All mice were imaged using a cooled CCD camera (Xenogen IVIS® 200 Series), using Live Imaging® software for image acquisition and analysis (Caliper Life Science, Hopkinton, MA). Color-coded whole-body images were recorded on days 0, 4, 7, 8, 11, 14, 17, 21, and 35 after the first treatment to measure the dynamics of peritoneal tumor growth. In these experiments, all images were acquired and collected using identical system settings: exposure time = 1 sec; binning = medium; f/stop 2 for ES-2-luc-bearing animals and exposure time = 10 sec; binning = medium; f/stop 2 for SK-OV-3-luc-bearing animals. D-luciferin (Caliper Life Science, Hopkinton, MA) at 113 mg/kg was injected IP into ES-2-luc-bearing and SK-OV-3-luc-bearing mice 5 and 20 min, respectively, prior to whole-body imaging. Live Imaging® software was used to quantify the total photon counts of bioluminescence from ES-2-luc-bearing or SK-OV-3-luc cells in the regions of interest (ROIs), which was selected manually over IP tumors.
2.2.6. Whole-body microPET/CT imaging
The proliferation of ES-2-luc cells was noninvasively monitored in vivo, using an Inveon microPET/CT hybrid scanner (Siemens, Knoxvile, TN). Animals were intraocularly (IO) injected with 107 ± 10 μCi of 18F-FLT on days 0, 7, and 17 after the first treatment. One hour after injection, mice were prepared for dual hybrid microPET/CT imaging; microPET acquisition was performed immediately, followed by microCT acquisition. Maximum intensity projections were created using a Siemens Inveon Research Workplace (Knoxville, TN); PET images were reconstructed using OSEM3D/MAP. MicroCT images were also reconstructed using standard conebeam reconstruction. Both microPET and microCT images were fused in Siemens Inveon Software. ROIs were manually drawn on CT pictures around the region with highest signal intensity in the IP cavity, and tumor volume and 18F-FLT uptake, assessed by standard uptake value (SUV) mean, were calculated by the estimation of voxels within tomographic planes. The relative SUV (% SUV) was calculated by comparing the SUV mean at each time point to the initial SUV mean on day 0.
2.2.7. Statistical analysis
Data were represented as mean ± standard deviation (SD). Statistical analysis was conducted using one-way ANOVA at 5% significance level combined with Tukey’s multiple comparison tests provided by GraphPad Prism. (*), (**), and (***) signify p < 0.05, p < 0.01, and p < 0.001, respectively.
3. Results
3.1. Characterization of drug-loaded PEG-b-PCL micelles
Table 1 presents the level of PTX, CYP, and GSP in 0.9% saline (mg/mL) and loading efficiency (weight drug/weight polymer), achieved after loading in PEG-b-PCL micelles by a solvent evaporation technique. PEG-b-PCL micelles had z-average diameter of 80–90 nm and PDI < 0.1 regardless of drug loading (supplementary material). The maximum loading efficiency of PTX, CYP, and GSP in PEG-b-PCL micelles was ca. 2, 1, and 1%, respectively. Similarly, 2-drug PEG-b-PCL micelles had low loading efficacy (Table 1). In both cases at 200 mg/mL of PEG-b-PCL micelles, however, aqueous drug solubility was 0.9 to 3.8 mg/mL, which is enough to enable in vivo treatment studies. Interestingly, 3-drug PEG-b-PCL micelles had the highest drug loading efficiency, ca. 9.4%, achieving water solubility for PTX, CYP, and GSP at ca. 6 mg/mL of each drug for a total water solubility of 18 mg/mL.
Table 1.
Combination drug solubilization by PEG-b-PCL micelles.
| Micelles | Polymer (mg/mL) | PTX (mg/mL) | CVP (mg/mL) | GSP (mg/mL) | Loading efficiency (%) |
|---|---|---|---|---|---|
| 1-in-1 | 200 | 3.8 ± 0.6 | - | - | 2.0 ± 0.3 |
| 200 | - | 1.2 ± 0.1 | - | 0.6 ± 0.1 | |
| 200 | - | - | 2.3 ± 0.5 | 1.2 ± 0.3 | |
| 2-in-1 | 200 | 3.6 ± 0.4 | 2.7 ± 0.5 | 3.1 ± 0.5 | |
| 200 | 2.8 ± 0.2 | 0.9 ± 0.0 | 1.8 ± 0.1 | ||
| 200 | 3.2 ± 1.0 | 3.2 ± 0.2 | 3.2 ± 0.6 | ||
| 3-in-1 | 200 | 6.3 ± 0.5 | 6.2 ± 0.5 | 6.2 ± 0.5 | 9.4 ± 0.8 |
After 24 h at 25 °C, single drug PEG-b-PCL micelles retained CYP and GSP, ca. 96 and 89%, respectively, whereas PEG-b-PCL micelles retained 34% of PTX, indicating poor physical stability (supplementary material). Not surprisingly, 2-drug PEG-b-PCL micelles also poorly retained PTX, and PEG-b-PCL micelles containing CYP and GSP were stable, ca. 91 and 90%, respectively. 3-drug PEG-b-PCL micelles showed anomalous stability, retaining PTX, CYP, and GSP at ca. 99%, 37%, and 82%, respectively. A similar relative pattern of in vitro drug release for 3-drug PEG-b-PCL micelles was observed (Fig. 2). All three drugs were released from PEG-b-PCL micelles over 72 h in a biphasic pattern, and the individual release curves were fit to a two-phase exponential association model with the goodness of fit of 0.98 (PTX), 1.00 (CYP), and 0.99 (GSP) (Fig. 2). The t1/2 values of PTX, CYP, and GSP for PEG-b-PCL micelles were 50, 18, and 30 h, respectively. Drug release curves reached a plateau at 60, 93, and 67% for PTX, CYP, and GSP, respectively, within 72 h.
Fig 2.
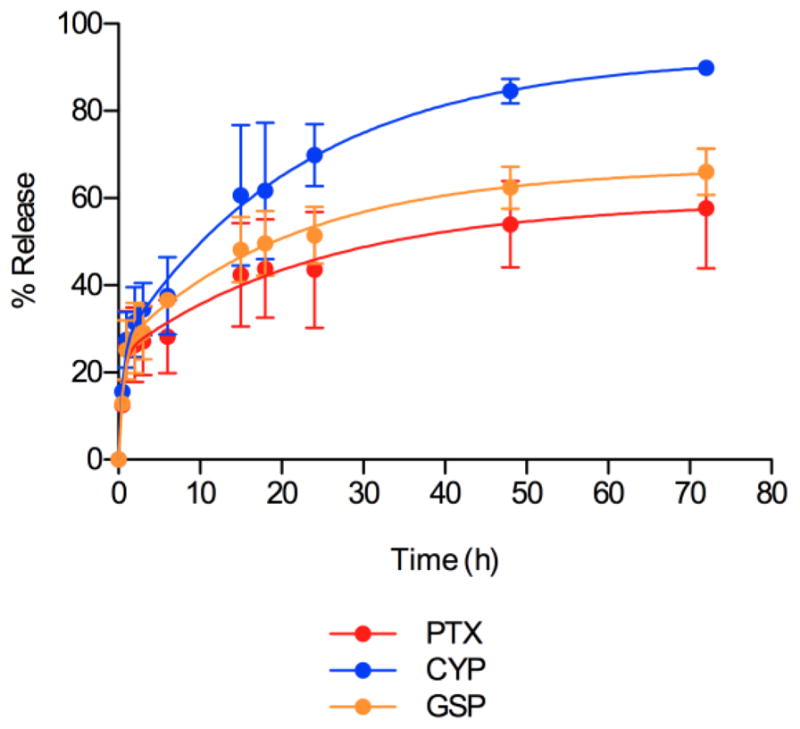
In vitro drug release kinetics of PTX, CYP, and GSP from PEG-b-PCL micelles.
3.2. In vitro cytotoxicity study
In 2-D and 3-D cell culture, PEG-b-PCL micelles containing PTX had the highest in vitro cytotoxicity against ES-2-luc and SKOV3-luc cells based on a loss of bioluminescence signal upon drug treatment (Table 2). CYP was also relatively active against luciferase-expressing ES-2-luc having IC50 values in 2-D and 3-D cell culture at 234 and 65 nM, respectively, whereas it was much less active against SKOV3-luc cells, IC50 > 100,000 nM. GSP as a single agent was the least active. Accordingly, 2-drug PEG-b-PCL micelles containing PTX/CYP and PTX/GSP were relatively active against ES-2-luc and SKOV3-luc cells, whereas 2-drug PEG-b-PCL micelles containing CYP/GSP were largely ineffective. The IC50 of 3-drug PEG-b-PCL micelles was 51, 101, and 67 nM for ES-2-luc (2-D), ES-2-luc (3-D), and SKOV3-luc (2-D), respectively. Morphologies of ES-2-luc tumor spheroids upon drug treatment at 1 μM were largely consistent with IC50 values for ES-2-luc cells in 3-D cell culture (Fig. 3): PTX had the greatest impact on the growth of ES-2-luc tumor spheroids, followed by CYP; GSP did not appear to restrict the growth of ES-2-luc tumor spheroids. In all, single drug and 2-drug treatments, ES-2-luc tumor spheroids retained a spherical morphology. In contrast, ES-2-luc tumor spheroids lost their integrity after treatment with 3-drug PEG-b-PCL micelles, appearing to disaggregate into smaller cellular aggregates that lack a defined morphology (Fig. 3).
Table 2.
IC50 values of PTX, CYP, and GSP against 2-D- and 3-D- ES-2-luc- and 2-D- SKOV3-luc cells based on bioluminescence imaging.
| Cells (Condition) | PTX (nM) | CYP (nM) | GSP (nM) | PTX/CYP (nM) (1:2 molar ratio) | CYP/GSP (nM) (1:1) | GSP/PTX (nM) (2:1) | PTX/CYP/GSP (nM) (1:2:2) |
|---|---|---|---|---|---|---|---|
| ES-2 (2D) | 12 ± 5 | 240 ± 54 | >100000 | 27 ± 4 | 18790+4119 | 40 ± 15 | 51 ± 18 |
| ES-2 (3D) | 9 ± 3 | 65 ± 10 | 4823 ± 1521 | 62 ± 14 | 2550 ± 920 | 108 ± 22 | 101 ± 21 |
| SKOV3 (2D) | 98 ± 15 | >100000 | >100000 | 58 ± 11 | >100000 | 67 ± 11 | 67 ± 35 |
Fig 3.
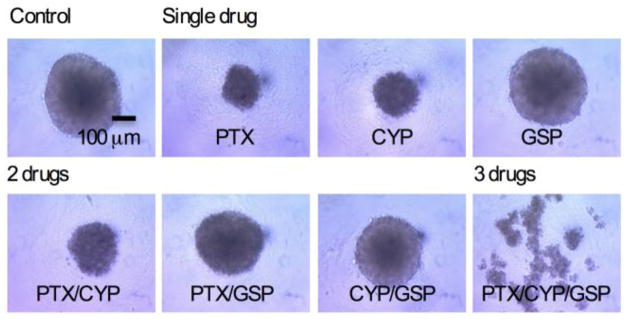
Morphologies of ES-2-luc spheroids 3 days post treatment: Vehicle (control), single drug, 2-drug, and 3-drug. ES-2-luc spheroids were treated with 1 μM of total drug(s) for each formulation.
3.3. Anticancer efficacy of 3-drug PEG-b-PCL micelles after IP injection
The acute toxicity of 3-drug PEG-b-PCL micelles was assessed in female 6–8 week-old athymic nude-Foxn1nu mice, following IP injection (q7d × 3), monitoring body weight, general appearance, and mortality. 3-drug PEG-b-PCL micelles at 30/30/30 mg/kg and PEG-b-PCL micelles containing only PTX at 50 mg/kg were well tolerated in a q7d × 3 schedule over 40 days (supplementary material).
Peritoneal dissemination of ES-2-luc cells was observed longitudinally in groups of four mice, monitoring bioluminescence relative to initial signal (%BLI) over the peritoneal cavity as an signal of tumor burden (Figs. 4A and 4B). By day 4 after IP inoculation of ES-2-luc cells, regional bioluminescence from the peritoneal cavity of mice suggested that ES-2-luc cells had already formed solid tumors (Fig. 4A) [17, 18]. In a vehicle control, bioluminescence from ES-2-luc cells in the peritoneum increased rapidly over three weeks, and ascites, i.e. fluid in peritoneum, rapidly formed in mice based on noninvasive bioluminescence images fused with white-light pictures (Figs. 4A and 4F). Twenty-one days after control treatment (25 days post cell inoculation), bioluminescence signals of non-treated ES-2-luc-bearing mice increased 8-fold (Fig. 4B), and the average abdominal radius of control mice increased 1.2-fold due to ascite formation (Fig. 4C). Tumor burden killed control mice within 27 days post cell inoculation (Fig. 4E); ca. 4 mL of ascites was in the peritoneum. Extensive metastatic tumor foci were observed adhered onto stomach, intestines, colon, and kidneys in sacrificed animals (data not shown).
Fig 4.

Nonevasive bioluminescence imaging and treatment assessment for metastatic ES-2-luc ovarian xenograft model. Results are represented by whole-body bioluminescence images of mice obtained with IVIS imaging system (A), quantitative bioluminescence intensity (BLI) in ROIs (B), and % change of radius of abdomen of mice (C). Body weight change (D), Kaplan-Meier analysis for survival (E), and white light observation (on day 25 post cell inoculation) (F) of tumor-bearing mice receiving IP treatment of vehicle, 3-drug PEG-b-PCL micelles at 30, 30, and 30 mg/kg, or PEG-b-PCL micelles containing PTX at 30 mg/kg on a q7d × 3 schedule (* <0.05, ** <0.01, *** <0.001).
PEG-b-PCL micelles containing PTX (30 mg/kg) were effective in delaying tumor progression during drug treatment (Figs. 4A and 4B); however, %BLI increased rapidly after termination of drug treatment, indicating regrowth of tumors. In this treatment group, we observed less ascite formation (Figs. 4A and 4F). After 21 days, %BLI was 3-fold higher than the start of treatment and 2.8-fold less than vehicle control mice (Fig. 4B); however, all mice died within 35 days post cell inoculation (Fig. 4E).
3-drug PEG-b-PCL micelles with PTX, CYP, and GSP at 30, 30, 30 mg/kg, respectively, also prevented tumor progression (Figs. 4A and 4B), but notably, %BLI did not increase after the cessation of treatment. Within the first week after treatment, tumors seemed to develop faster than tumors in PTX-treated mice. But after the second dose, %BLI decreased and reached a plateau for about two weeks. After 21 days, the %BLI value for 3-drug PEG-b-PCL micelles was 11-fold and 4-fold less than the vehicle control and PTX, respectively (Fig. 4B). The average radius of abdomen did not significantly change over time (Fig. 4C). Lastly, 50% of animals treated with 3-drug PEG-b-PCL micelles survived for 46 days post cell inoculation.
To validate in vivo results from bioluminescence imaging, 18F-FLT was used as a PET tracer for the noninvasive visualization of cell proliferation of IP injected ES-2-luc cells and assessment of treatment responses (Fig. 5A) [19–22]. In the vehicle control, PET imaging showed large tumor masses (highlighted in yellow) at 4 days post cell inoculation that grew larger over 17 days. Tumors were also observed at the start of treatment for PTX-treated mice, but they did not appear to change in size during the course of treatment. In contrast, while tumors were readily discerned at the start of treatment tumors treated with 3-drug PEG-b-PCL micelles were eradicated by day 17, noting instead a diffuse signal from ES-2 cells throughout the peritoneum (Fig 5A). The high signal of 18F-FLT in the posterior region of mice is due to uptake of 18F-FLT by the kidneys. %SUV on day 17, as a measure of uptake of 18F-FLT in ES-2-luc tumors, was 263, 164, and 86% for vehicle, PTX, and 3-drug PEG-b-PCL micelles, respectively (Fig. 5B).
Fig 5.

Nonevasive microPET/CT imaging and treatment assessment for metastatic ES-2-luc ovarian xenograft model. Results are represented by whole-body images (A), tracer uptake in ROIs expressed as % SUV (B) of tumor-bearing mice receiving IP treatment of vehicle, 3-drug PEG-b-PCL micelles at 30, 30, and 30 mg/kg, or PEG-b-PCL micelles containing PTX at 30 mg/kg on a q7d × 3 schedule (* <0.05, ** <0.01, *** <0.001). Tumor tissues at day 0 reached 100–200 mm3 in volume.
Additionally, 3-drug PEG-b-PCL micelles with PTX, CYP, and GSP at 30, 30, 30 mg/kg, respectively, was tested in a SKOV3-luc ovarian cancer xenograft model of peritoneal carcinomatosis (Fig. 6). In comparison to the ES-2-luc xenograft model, growth of SKOV3-luc cells in the peritoneum of mice was slower, and treatment was started 16 days post cell inoculation after appearance of a bioluminescence signal. For the vehicle control, %BLI grew slowly over two weeks and then grew more rapidly for the next two weeks. Both treatment groups were very effective against SKOV3-luc ovarian cancer (Figs. 6A and 6B), causing a slight decrease in %BLI: 3-fold and 1.5-fold for 3-drug and PTX-treated mice. After 35 days (51 days post cell inoculation), %BLI for 3-drug PEG-b-PCL micelles was 2-fold and 73-fold less than %BLI for PTX-treated mice and vehicle control, respectively. Lastly, 75% of vehicle control and 25 % of PTX-treated mice died within 58 days post cell inoculation; however, 100% of the animals receiving 3-drug PEG-b-PCL micelles survived with < 15% body weight change until the termination of experiments (Figs. 6C, 6D, and 6E).
Fig 6.

Nonevasive bioluminescence imaging and treatment assessment for metastatic SKOV3-luc ovarian xenograft model Results are represented by whole-body bioluminescence images of mice obtained with IVIS imaging system (A) and quantitative bioluminescence intensity (BLI) in ROIs (B). Body weight change (C), Kaplan-Meier analysis for survival (D), and white light observation (on day 45 post cell inoculation) (E) of tumor-bearing mice receiving IP treatment of vehicle, 3-drug PEG-b-PCL micelles at 30, 30, and 30 mg/kg, or PEG-b-PCL micelles containing PTX at 30 mg/kg on a q7d × 3 schedule (* <0.05, ** <0.01, *** <0.001).
4. Discussion
IP drug delivery for treatment of peritoneal metastases of ovarian cancer has produced survival benefits in multiple clinical trials despite the fact that IP treatments rely chiefly on vehicles that were developed for intravenous (IV) drug delivery, e.g. Cremophor EL for PTX [10]. More recently, novel drug delivery systems for IP drug delivery, e.g. liposomes, micro-particles, and sustained release gels have generated promising results in ovarian xenograft models [23]. However, research on IP drug delivery has largely focused on the delivery of a single cytotoxic agent. Less attention has been devoted to the IP delivery of molecularly targeted agents for the treatment of ovarian cancer and even less attention on the IP delivery of drug combinations of cytotoxic agent and molecularly targeted agent(s). Amiji and coworkers have done early studies on combination drug delivery using biodegradable polymeric nanoparticles containing PTX and ceramide or PTX and tamoxifen, overcoming multidrug resistance in subcutaneous SKOV3 xenograft models [24, 25].
Multi-drug loaded PEG-b-PCL micelles may potentially fulfill several major requirements for IP combination drug delivery (Fig. 1): biocompatibility, multiple drug solubilization, physical stability against drug precipitation, and sustained release. Multi-drug loaded PEG-b-PCL micelles enable multi-drug treatment after a single IP injection or infusion, nullifying the requirement of sequential IP dosing of drug combinations for the treatment of ovarian cancer. Multi-drug loaded PEG-b-PCL micelles may derive a major safety benefit for IP combination drug delivery by the elimination of multiple vehicles, e.g. Cremophor EL, co-solvents, which are often toxic after either IV or IP administration, noting that the maximum tolerable dose (MTD) of IP bolus PTX solubilized by Cremophor EL was 20 mg/kg/week [26].
PEG-b-PCL micelles prepared by a solvent evaporation technique had a good capacity for PTX, CYP, and GSP, ca. 9.4%, resulting in multiple drug solubilization at 18 mg/mL, ca. 6.2 mg/mL for each anticancer agent, and PEG-b-PCL micelles were obtained with z-average diameters at ca. 80 nm, regardless of drug loading (Table 1). In prior preclinical and clinical studies, PTX and GSP were solubilized by CrEL and PEG400/Cremophor EL, respectively, for IV administration, and CYP was chemically-modified into a water-soluble analogue (IPI-926) [13, 27, 28]. 3-drug PEG-b-PCL micelles were fairly physically stable at 25 °C, although ca. 63% of CYP was lost due to precipitation over a 24 h period (supplementary material). For in vitro and in vivo studies, 3-drug PEG-b-PCL micelles were tested within ca 1–2 h after preparation or stored at 4 °C, where there was higher physical stability (data not shown). It is noted that drug loss due to precipitation has also been observed for PTX in PEG-b-PLA micelles (Genexol-PM) at 25 °C over 24 h. Freeze-drying of Genexol-PM and reconstitution were used to enable a stable IV administrated PTX in a sterile aqueous vehicle for clinical trials.
In vitro evidence for sustained release of PTX, CYP, and GSP from 3-drug PEG-b-PCL micelles was obtained under approximate sink conditions, with calculated t1/2 values at 50, 18, and 30 h, respectively (Fig. 2). Our prior studies on 3-drug poly(ethylene glycol)-block-poly(d,l-lactic acid) micelles showed a correlation between the t1/2 values for in vitro multi-drug release and octanol-water partition coefficients (log P) [29]; In contrast, an opposite trend was observed for 3-drug PEG-b-PCL micelles with PTX, CYP, and GSP, with CYP having the fastest release and highest log P value, ca. 6.1. The relatively fast release of CYP from 3-drug PEG-b-PCL micelles was consistent with the loss of CYP due to precipitation over a 24 h period at 25 °C (supplementary material). Mechanistic analysis for 3-drug PEG-b-PCL micelles merits attention, focusing on regions of drug solubilization in PEG-b-PCL micelles and drug-drug interactions. In summary, 3-drug PEG-b-PCL micelles release PTX, CYP, and GSP over a few days in vitro; after IP injection we also predict sustained multi-drug release, which will be verified in pharmacokinetic experiments.
The in vitro and in vivo activity of 3-drug PEG-b-PCL micelles was tested against two human ovarian cancer cell lines, owing to four histological subtypes of ovarian cancer: Serous, mucinous, clear cell, and endometrioid; and tumors having no-distinctive histological features are designated as undifferentiated adenocarcinoma [30]. ES-2 is categorized as undifferentiated adenocarcinoma (aggressive subtype), and SKOV3 is categorized in serous adenocarcinoma (moderate-grade subtype) [31]. 3-drug PEG-b-PCL micelles had IC50 values of 51 and 67 nM against ES-2-luc and SKOV3-luc ovarian cancer cells, respectively, based on a decline in bioluminescence, reflecting decreased protein expression (luciferase) (Table 2). Previous work by De Souza et al. showed a linear correlation between bioluminescence from SKOV3-luc cells and cell viability based on the MTT assay [8]. While the activity of 3-drug PEG-b-PCL micelles was not better than PTX in 2-D and 3-D cell culture in terms of IC50 value, 3-drug PEG-b-PCL micelles at 1000 nM were unique in the disaggregation of ES-2-luc spheroids into smaller cellular aggregates, whereas ES-2-luc spheroids stayed intact after treatment with PTX, but with reduced growth of tumor spheroids, presumably by acting of proliferating ES-2-luc cells in the periphery. It is noted that ovarian cancer cells show anchorage-independent growth in ascites by the formation of tumor spheroids and that ovarian tumor spheroids have been linked to drug resistance due to poor drug penetration or increased pro-survival signaling. Brown and coworkers have also shown that compact ES-2 tumor spheroids have an invasive phenotype, whereas loose, sheet-like SKOV-3 aggregates were unable to invade a 3-D collagen matrix in vitro [32]. Thus, disaggregation of ES-2-luc spheroids by 3-drug PEG-b-PCL micelles may enhance drug delivery to ovarian cancer cells and preclude invasive behavior, resulting in improved treatment outcomes. It is speculated that disaggregation of ES-2-luc spheroids is induced by multiple mechanisms, such as inhibition of Hh signaling by CYP possibly reversing taxane resistance, improved penetration of PTX into spheroids, and GSP augmented apoptosis.
Both bioluminescence imaging and microPET/CT imaging showed that PTX, CYP, and GSP, injected IP as 3-drug PEG-b-PCL micelles, has potent antitumor activity in metastatic ES-2-luc and SKOV3-luc xenograft models (Figs. 4, 5, and 6). At the start of drug treatment (4 days post cell inoculation), ES-2-luc tumors were plainly visible in the peritoneum by microPET/CT imaging along with extensive ascite formation, suggesting an advanced stage of disease. Bioluminescence imaging and microPET/CT imaging showed that the tumors spread rapidly in the peritoneum, and median survival time < 30 days. The lethality of ES-2 cells in an IP metastatic xenograft model has been noted in the literature [33]. 3-drug PEG-b-PCL micelles with PTX, CYP, and GSP at 30, 30, 30 mg/kg, respectively, prevented the metastatic spread of ES-2-luc tumors in the peritoneum and ascite formation (Fig. 4). Furthermore, ES-2-luc tumors, plainly visible at the onset of treatment, were completely eradicated after treatment with 3-drug PEG-b-PCL micelles (q7d × 3), based on microPET/CT imaging using 18F-FLT as a PET tracer (Fig. 5A). Accordingly, ES-2-luc-bearing mice survived longer than mice treated with PTX, where ES-2-luc tumors were still visible after drug treatment (q7d × 3). The antitumor efficacy of 3-drug PEG-b-PCL micelles with PTX, CYP, and GSP was also confirmed in an IP metastatic SKOV3-luc xenograft model (Fig. 6). In summary, 3-drug PEG-b-PCL micelles disassembled ES-2-luc tumor spheroids in vitro, eradicated metastatic ES-2-luc tumors in vivo, and prolonged survival, validating the IP delivery of multi-drug polymeric micelles as a meaningful drug delivery strategy for the treatment of ovarian cancer.
5. Conclusions
IP drug delivery of PTX, CYP, and GSP was enabled by PEG-b-PCL micelles, fulfilling several major requirements for combination drug delivery: Biocompatibility, multiple drug solubilization, physical stability against drug precipitation, and sustained release. Multi-drug loaded PEG-b-PCL micelles prepared by a simple solvent evaporation technique enabled solubilization of PTX, CYP, and GSP at ca. 18 mg/mL. PEG-b-PCL micelles carrying PTX, CYP, and GSP were nanoscopic, fairly stable in aqueous solution, and capable of simultaneous sustained release of PTX, CYP, and GSP in vitro. This 3-drug combination was highly effective in metastatic ES-2-luc and SKOV3-luc xenograft models, eradicating peritoneal tumors and prolonging survival over PTX alone. Antitumor efficacy of PTX, CYP, and GSP may partly be explained by their ability to disassemble ovarian tumor spheroids, which are drug resistant. These results clearly show the unique potential of multi-drug delivery via IP-injected polymeric micelles for the treatment of metastatic ovarian cancer. Future studies will focus on the pharmacokinetic/pharmacodynamic relationships of IP-administered drug combinations in metastatic ovarian xenograft models.
Supplementary Material
Acknowledgments
This work was financially supported by National Institutes of Health (R21 CA-161537) and Carbone Cancer Center at University of Wisconsin-Madison. Authors extend their gratitude to Small Imaging Animal Facility in the Carbone Comprehensive Cancer Center, University of Wisconsin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA: a cancer journal for clinicians. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Harries M, Gore M. Part II: chemotherapy for epithelial ovarian cancer-treatment of recurrent disease. The lancet oncology. 2002;3:537–545. doi: 10.1016/s1470-2045(02)00847-1. [DOI] [PubMed] [Google Scholar]
- 3.Ray A, Meng E, Reed E, Shevde LA, Rocconi RP. Hedgehog signaling pathway regulates the growth of ovarian cancer spheroid forming cells. International journal of oncology. 2011;39:797–804. doi: 10.3892/ijo.2011.1093. [DOI] [PubMed] [Google Scholar]
- 4.Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM, Alvarez RD, Landen CN. Smoothened antagonists reverse taxane resistance in ovarian cancer. Molecular cancer therapeutics. 2012;11:1587–1597. doi: 10.1158/1535-7163.MCT-11-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Zhou JY, Zhang L, Wu GS. Involvement of MKP-1 and Bcl-2 in acquired cisplatin resistance in ovarian cancer cells. Cell Cycle. 2009;8:3191–3198. doi: 10.4161/cc.8.19.9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bajaj G, Yeo Y. Drug delivery systems for intraperitoneal therapy. Pharmaceutical research. 2010;27:735–738. doi: 10.1007/s11095-009-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbolina MV, Moss NM, Westfall SD, Liu Y, Burkhalter RJ, Marga F, Forgacs G, Hudson LG, Stack MS. Microenvironmental regulation of ovarian cancer metastasis. Cancer treatment and research. 2009;149:319–334. doi: 10.1007/978-0-387-98094-2_15. [DOI] [PubMed] [Google Scholar]
- 8.De Souza R, Zahedi P, Badame RM, Allen C, Piquette-Miller M. Chemotherapy dosing schedule influences drug resistance development in ovarian cancer. Molecular cancer therapeutics. 2011;10:1289–1299. doi: 10.1158/1535-7163.MCT-11-0058. [DOI] [PubMed] [Google Scholar]
- 9.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. The Journal of pharmacology and experimental therapeutics. 2007;322:80–88. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 10.Lu Z, Wang J, Wientjes MG, Au JL. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010;6:1625–1641. doi: 10.2217/fon.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes & development. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YW, Wang LS, Chang HL, Ye W, Dowd MK, Wan PJ, Lin YC. Molecular mechanisms of (−)-gossypol-induced apoptosis in human prostate cancer cells. Anticancer research. 2006;26:1925–1933. [PubMed] [Google Scholar]
- 13.Jia L, Coward LC, Kerstner-Wood CD, Cork RL, Gorman GS, Noker PE, Kitada S, Pellecchia M, Reed JC. Comparison of pharmacokinetic and metabolic profiling among gossypol, apogossypol and apogossypol hexaacetate. Cancer chemotherapy and pharmacology. 2008;61:63–73. doi: 10.1007/s00280-007-0446-3. [DOI] [PubMed] [Google Scholar]
- 14.Hasenstein JR, Shin HC, Kasmerchak K, Buehler D, Kwon GS, Kozak KR. Anti-Tumor Activity of Triolimus: A Novel Multi-Drug Loaded Micelle Containing Paclitaxel, Rapamycin and 17-Aag. Molecular cancer therapeutics. 2012 doi: 10.1158/1535-7163.MCT-11-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho H, Kwon GS. Polymeric micelles for neoadjuvant cancer therapy and tumor-primed optical imaging. ACS nano. 2011;5:8721–8729. doi: 10.1021/nn202676u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Spheroid-based drug screen: considerations and practical approach. Nature protocols. 2009;4:309–324. doi: 10.1038/nprot.2008.226. [DOI] [PubMed] [Google Scholar]
- 17.Zinn KR, Chaudhuri TR, Szafran AA, O’Quinn D, Weaver C, Dugger K, Lamar D, Kesterson RA, Wang X, Frank SJ. Noninvasive bioluminescence imaging in small animals, ILAR journal/National Research Council. Institute of Laboratory Animal Resources. 2008;49:103–115. doi: 10.1093/ilar.49.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dufort S, Sancey L, Wenk C, Josserand V, Coll JL. Optical small animal imaging in the drug discovery process. Biochimica et biophysica acta. 2010;1798:2266–2273. doi: 10.1016/j.bbamem.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 19.Lee HJ, Tantawy MN, Nam KT, Choi I, Peterson TE, Price RR. Evaluation of an intraperitoneal ovarian cancer syngeneic mouse model using 18F-FDG MicroPET imaging. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2011;21:22–27. doi: 10.1097/IGC.0b013e3182021bda. [DOI] [PubMed] [Google Scholar]
- 20.Jensen MM, Erichsen KD, Bjorkling F, Madsen J, Jensen PB, Hojgaard L, Sehested M, Kjaer A. Early detection of response to experimental chemotherapeutic Top216 with [18F]FLT and [18F]FDG PET in human ovary cancer xenografts in mice. PloS one. 2010;5:e12965. doi: 10.1371/journal.pone.0012965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aide N, Kinross K, Cullinane C, Roselt P, Waldeck K, Neels O, Dorow D, McArthur G, Hicks RJ. 18F-FLT PET as a surrogate marker of drug efficacy during mTOR inhibition by everolimus in a preclinical cisplatin-resistant ovarian tumor model. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2010;51:1559–1564. doi: 10.2967/jnumed.109.073288. [DOI] [PubMed] [Google Scholar]
- 22.Zavaleta CL, Phillips WT, Bradley YC, McManus LM, Jerabek PA, Goins BA. Characterization of an intraperitoneal ovarian cancer xenograft model in nude rats using noninvasive microPET imaging. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2007;17:407–417. doi: 10.1111/j.1525-1438.2007.00814.x. [DOI] [PubMed] [Google Scholar]
- 23.Vassileva V, Moriyama EH, De Souza R, Grant J, Allen CJ, Wilson BC, Piquette-Miller M. Efficacy assessment of sustained intraperitoneal paclitaxel therapy in a murine model of ovarian cancer using bioluminescent imaging. British journal of cancer. 2008;99:2037–2043. doi: 10.1038/sj.bjc.6604803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devalapally H, Duan Z, Seiden MV, Amiji MM. Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:3193–3203. doi: 10.1158/1078-0432.CCR-07-4973. [DOI] [PubMed] [Google Scholar]
- 25.Devalapally H, Duan Z, Seiden MV, Amiji MM. Paclitaxel and ceramide co-administration in biodegradable polymeric nanoparticulate delivery system to overcome drug resistance in ovarian cancer. International journal of cancer. Journal international du cancer. 2007;121:1830–1838. doi: 10.1002/ijc.22886. [DOI] [PubMed] [Google Scholar]
- 26.Vassileva V, Grant J, De Souza R, Allen C, Piquette-Miller M. Novel biocompatible intraperitoneal drug delivery system increases tolerability and therapeutic efficacy of paclitaxel in a human ovarian cancer xenograft model. Cancer chemotherapy and pharmacology. 2007;60:907–914. doi: 10.1007/s00280-007-0449-0. [DOI] [PubMed] [Google Scholar]
- 27.McCann CK, Growdon WB, Kulkarni-Datar K, Curley MD, Friel AM, Proctor JL, Sheikh H, Deyneko I, Ferguson JA, Vathipadiekal V, Birrer MJ, Borger DR, Mohapatra G, Zukerberg LR, Foster R, Macdougall JR, Rueda BR. Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PloS one. 2011;6:e28077. doi: 10.1371/journal.pone.0028077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terwogt JM, Nuijen B, Huinink WW, Beijnen JH. Alternative formulations of paclitaxel. Cancer treatment reviews. 1997;23:87–95. doi: 10.1016/s0305-7372(97)90022-0. [DOI] [PubMed] [Google Scholar]
- 29.Shin HC, Alani AW, Cho H, Bae Y, Kolesar JM, Kwon GS. A 3-in-1 polymeric micelle nanocontainer for poorly water-soluble drugs. Molecular pharmaceutics. 2011;8:1257–1265. doi: 10.1021/mp2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berns EM, Bowtell DD. The changing view of high-grade serous ovarian cancer. Cancer research. 2012;72:2701–2704. doi: 10.1158/0008-5472.CAN-11-3911. [DOI] [PubMed] [Google Scholar]
- 31.Lalwani N, Prasad SR, Vikram R, Shanbhogue AK, Huettner PC, Fasih N. Histologic, molecular, and cytogenetic features of ovarian cancers: implications for diagnosis and treatment. Radiographics : a review publication of the Radiological Society of North America, Inc. 2011;31:625–646. doi: 10.1148/rg.313105066. [DOI] [PubMed] [Google Scholar]
- 32.Sodek KL, Ringuette MJ, Brown TJ. Compact spheroid formation by ovarian cancer cells is associated with contractile behavior and an invasive phenotype. International journal of cancer. Journal international du cancer. 2009;124:2060–2070. doi: 10.1002/ijc.24188. [DOI] [PubMed] [Google Scholar]
- 33.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Molecular therapy : the journal of the American Society of Gene Therapy. 2004;10:1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.