

Abstract
The concept of eliminating HIV-1 infectivity by elevating the viral mutation rate was first proposed over a decade ago, even though the general concept had been conceived earlier for RNA viruses. Lethal mutagenesis was originally viewed as a novel chemotherapeutic approach for treating HIV-1 infection in which use of a viral mutagen would over multiple rounds of replication lead to the lethal accumulation of mutations, rendering the virus population non infectious – known as the slow mutation accumulation model. There have been limitations in obtaining good efficacy data with drug leads, leaving some doubt into clinical translation. More recent studies of the APOBEC3 proteins as well as new progress in the use of nucleoside analogs for inducing lethal mutagenesis have helped to refocus attention on rapid induction of HIV-1 lethal mutagenesis in a single or limited number of replication cycles leading to a rapid mutation accumulation model.
Keywords: retrovirus, evolution, quasispecies, hypermutation, error catastrophe, extinction catastrophe
Revisiting HIV-1 lethal mutagenesis
The concept of HIV-1 lethal mutagenesis (see Glossary) was first described over a decade ago and was received with tremendous promise and enthusiasm [1]. Over the intervening years, it has become clear that the details regarding the molecular basis for HIV-1 lethal mutagenesis are more complicated than was originally believed and that it has been quite challenging to identify therapeutics that would allow for translation of a clinically-approved lethal mutagenesis therapeutic strategy for HIV-1 infection. Important encouragement in development of lethal mutagenesis as an anti-HIV-1 strategy occurred in the past few years from the discovery of the APOBEC (apolipoprotein B mRNA editing complex) family of cytosine deaminases (particularly the APOBEC3 family), for which the prevailing evidence indicates that members of this family represent an evolutionarily conserved defense mechanism against viruses as well as other molecular parasites [2]. The primary antiviral activity of these enzymes is C-to-U hypermutagenesis of single-stranded viral DNAs – which for HIV-1 occurs during the minus-strand DNA synthesis of reverse transcription and leads to G-to-A hypermutagenesis in the plus-strand DNA [2]. The observation that cells have evolved antiviral mechanisms involving lethal mutagenesis suggests that therapeutic approaches involving enhanced viral mutagenesis are ultimately seeking to mimic what has already been established in evolutionary time as a potent and effective antiviral strategy.
The early thoughts regarding the basis of HIV-1 lethal mutagenesis – as well as approaches envisioned for other RNA viruses – proposed that the slow accumulation of mutations introduced by a mutagenic nucleotide analogue over multiple rounds of replication, which would slowly elevate the virus mutational load until the induction of lethal mutagenesis (known as the slow mutation accumulation model). The discovery of the APOBEC3 proteins led to the realization that lethal mutagenesis could arise in a much more robust and rapid manner. Specifically, APOBEC3 proteins can induce HIV-1 lethal mutagenesis within a single round of viral replication [2–4]. This observation is important because the rapid accumulation of mutations for inducing lethal mutagenesis helps to eliminate many concerns regarding the clinical translation of such an antiviral intervention strategy with the slow mutation accumulation model. One concern, which is not supported by experimental data, is the selection of new virus variants with altered biological properties. Here, we argue that both a new conceptual understanding of HIV-1 lethal mutagenesis as well as promising new experimental approaches to induce rapid virus extinction with minimal cell toxicity provide the basis for renewed enthusiasm in the development of this novel antiviral strategy.
Concept of lethal mutagenesis
The basic concept of lethal mutagenesis is based on the idea of reaching the error threshold as well as a collapse of viral infectivity among virus variants within the population (reviewed in [5–7]). This is a novel antiviral strategy for the elimination of virus infectivity by the elevation of viral mutations and has yet to be reached by clinically approved therapeutics, though the possibility of ribavirin acting as a mutagen in the treatment of viral infections (e.g., hepatitis C virus, HCV) cannot be excluded. Such an outcome could occur by virus replication in the presence of mutagenic agents such as nucleotide analogues, or could arise through the action of cellular nucleic acid editing enzymes, such as the APOBEC3 proteins (Figure 1). Holland and colleagues published the first experiments with poliovirus and vesicular stomatitis virus (VSV) to show that elevated viral mutagenesis led to a negative outcome in terms of the adaptation and survival of the virus population [8]. The term `lethal mutagenesis' was coined by Loeb, Mullins and colleagues [1] in the first published account of the error catastrophe concept in HIV-1. Error catastrophe is a term used to describe the scenario for when the consensus sequence within the viral population (called the `master' sequence) is no longer the most fit and there is a drifting into the sequence space that can lead to noninfectious and nonfunctional virus genome sequences [9]. The ultimate movement of change in the `master' sequence to an unfit viral genome leads to the successful penetration of the error threshold and entry into error catastrophe. Complete loss of the `master' sequence represents virus extinction.
Figure 1.
Concept of HIV-1 lethal mutagenesis (extinction catastrophe). The left and right y-axes describe the inverse relationship between mutation rate (μ) and viral infectivity, respectively. Viable virus populations exist below the virus extinction threshold. Small molecules (e.g., nucleotide analogs) and cellular proteins (e.g., APOBEC3G) can act as viral mutagens and increase the viral mutation rate (μ = 3.4 × 10−5 mutations/target base pair/replication cycle [75]), which is identified in the figure as the `optimal' mutation rate. An increase in mutation rate could result in either enhanced mutagenesis (including lethal defection, an intermediary step in the path towards virus extinction) or directly past the error threshold (dashed line) to induce extinction catastrophe, i.e., lethal mutagenesis. As the HIV-1 mutation rate is decreased through ablated mutagenesis (e.g., due to HIV-1 reverse transcriptase [RT] variants with higher fidelity), viral infectivity also diminishes, most likely due to replicative fitness costs.
Dapp et al
At the molecular level, lethal mutagenesis is initiated by an elevation of the viral mutation rate (i.e., enhanced mutagenesis). This increase in mutation rate can result in the generation of defective, but replication-competent viruses (called `defectors') that can interfere with the replication of non-defective virus, and therefore suppress overall viral replication [10–14]. The presence of viral `defectors' creates the situation of lethal defection (Figure 1), which is an intermediary step in the path towards lethal mutagenesis and ultimate virus extinction. Support for the lethal defection model comes from both experimental as well as computational data [10–14]. Continued viral mutagenesis during lethal defection leads to an intensification of lethal mutations that further extinguish viral infectivity. The kinetics for where, as well as how lethal mutations accumulate, is poorly understood, as are how these events interface with other viral and host-mediated mechanisms. An intriguing alternative to virus extinction via lethal mutagenesis is the reduction of viral infectivity by high replication fidelity, resulting in abated mutagenesis. In theory, prolonged replication at a low mutation rate would also result in virus extinction (Figure 1). This approach is similar to studies showing that high-fidelity viral mutants possess less virulence and may provide an alternative means for vaccine development [15, 16].
Foundational studies of lethal mutagenesis and slow mutation accumulation
Chemical mutagens can damage DNA in living systems and are generally considered both poorly tolerated and carcinogenic agents but some have been successfully developed into anti-tumor chemotherapeutics [17]. As therapeutics become more patient-specific and disease-specific, the era of broad-spectrum chemical mutagen therapy has been reinvigorated by the study of lethal mutagenesis with viruses. Provided a high enough mutational burden, an irreversible genetic meltdown of the entire population causes its elimination [18]. Fortunately, the immense population size, genome structure, and adaptive requirements of most virus populations have positioned them very near the extinction threshold. Evidence for this first emerged in picornavirus studies where a mere twofold increase in viral mutation frequency was required to reduce viral titers by 99% [8]. Lethal mutagenesis has since been observed in many other RNA virus systems, including: HIV-1 [19, 20], poliovirus [21, 22], foot and mouth disease virus (FMDV) [23], and lymphochoriomeningitis virus (LCMV) [24]. Evidence in cell culture systems indicate that the clinically approved ribavirin regimen, used to treat HCV since 1998, elicits antiviral activity via lethal mutagenesis though this mechanism has not been fully established in vivo [25, 26]. These early studies provided evidence for lethal mutagenesis, but less attention was focused on potential mechanisms. The early HIV-1 studies with 5-hydroxydeoxycytidine suggested the slow accumulation model (Figure 2), whereby lethal mutagenesis occurred by a slow accumulation of mutations over multiple rounds of replication, ultimately causing virus extinction [1]. By analogy, many of the other lethal mutagenesis studies – even if not directly stating such a model – inferred this type of model for mutation accumulation leading to high mutational loads and eventual lethal mutagenesis.
Figure 2.
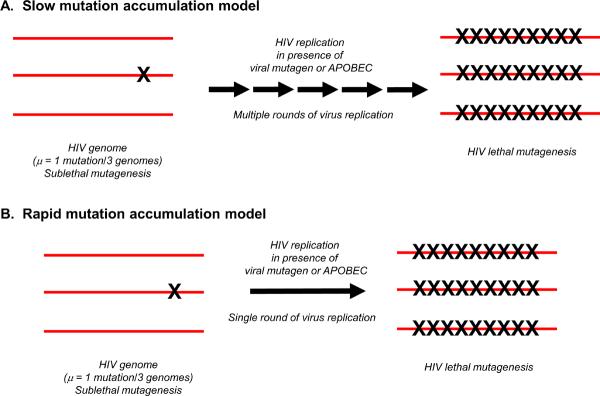
Mutation accumulation models for induction of HIV-1 lethal mutagenesis. (a) Slow mutation accumulation model. In this model, the increase in HIV-1 mutational load that results lethal mutagenesis is the result of a graduate increase in mutational load over multiple rounds of viral replication. (b) Rapid mutation accumulation model. In this model the increase in HIV-1 mutational load via lethal mutagenesis occurs rapidly, over a single or a few rounds of viral replication.
Dapp et al
Cell-culture based studies have shown promise for several viral systems, but the demonstration of lethal mutagenesis in vivo has been elusive. For example, phase II clinical trial results for KP-1461, a prodrug of KP-1212 (5,6-dihydro-5-aza-2'-deoxycytidine), a first-in-class viral mutagen for HIV-1, demonstrated there was perturbation of the mutation spectra, but no reduction in viral loads [27]. In particular, deep-sequence analysis of virus in these subjects revealed differences in the virus mutation spectra, but drug-exposed subjects showed no detectable differences in viral loads or CD4+ T cell counts, which are key prognosis markers for the level of HIV-1 infection. While retroviral recombination occurs at a high rate and could attenuate mutational burden among replication-competent virus, it is unlikely that it would prevent lethal mutagenesis or prevent any changes in viral loads or CD4+ T cell counts. One question that arises from these studies is whether KP-1461 could be effective as a monotherapy [27, 28], especially since the combined triple-therapy drug cocktail approach has been the standard for HIV-1-chemotherapy since the mid-1990s. Also, the clinically approved ribavirin regimen for treatment of HCV is administered in combination with interferon [29]. This suggests that the antiviral effect of KP-1461 may be enhanced in the presence of other antiviral agents. While monotherapy drug exposure typically leads to the rapid development of drug resistance, it is of note that there are no publications that document drug resistance to KP-1461. Taken together, these observations indicate that additional work is needed to document lethal mutagenesis of viruses in vivo.
HIV-1 lethal mutagenesis: rapid escalation in mutational load
As indicated above, the mechanism(s) of lethal mutagenesis had either directly stated (in the case of HIV-1) or had inferred (with other RNA viruses) the slow mutation accumulation model, where mutation accumulation over multiple rounds of replication was the envisioned mechanism (Figure 2). The discovery of the APOBEC3 proteins suggested that elevation of mutational load could occur rapidly, over a single or limited number of replication cycles. While early studies with HIV-1 used small molecules that could have been used to induce lethal mutagenesis in a rapid manner, concerns over the cytotoxicity of such molecules precluded intensive studies [1].
Lethal mutagenesis can be induced not only by drugs, but also by the human APOBEC3 family of proteins [2–4, 30–36]. The mechanism by which APOBEC3 proteins, the prototype being APOBEC3G (A3G), hypermutate retroviral genomes has been well-established [reviewed in [37], [38], and [39]]. Briefly, A3G is packaged into budding virions, after which the virion matures and binds to a target cell. In the target cell, A3G deaminates cytosines present in the minus-strand viral DNA during reverse transcription process. The deamination of cytosine to uracil causes a pre-mutatgenic lesion. This site templates for adenine during plus strand DNA synthesis rather than guanine. Deamination of cytosine by A3G during reverse transcription generates G-to-A mutation signatures in the resulting provirus [2]. However, its ability to mutate the viral genome depends on how well it can overcome HIV-1 viral infectivity factor (Vif) accessory protein. In a host specific manner, Vif targets A3G proteins for proteosomal degradation. By saturating A3G levels or less-stringent Vif alleles, A3G proteins can gain access to the nascent virions and mutate the viral genome as described above. An A3G deaminase-independent mechanism has been proposed as a major antiviral mechanism of action for HIV-1, however, this model remains controversial [40–43].
Cell culture work convincingly shows that these mutagen-inducing proteins can dispose of viral genomes in short order, i.e., over 90% loss in viral infection within a single replication cycle [2–4]. Because of the processivity of A3G once it engages the viral genome, evidence suggests the mutagenesis is a discrete all-or-nothing process [44, 45]. However, some studies suggest that A3G can contribute to sublethal mutagenesis [46], which include shaping the emergence of HIV-1 drug resistance [47–49]. Currently, the extent to which APOBEC3 proteins have contributed HIV-1 evolution remains controversial [50]; although, it is clear that HIV-1-infected patient samples reveal that APOBEC3 -signature mutations are actively found [51, 52].
A3G and A3F remain at the center of much recent focus as powerful HIV-1 mutators [reviewed [53] and [54]]. Both A3G and A3F, along with A3B, possess the capacity to restrict other retroviral genera as well, including: the murine leukemia virus (MLV, gammaretrovirus) [2, 3, 33], human T-lymphotropic virus 1 (HTLV-1, deltaretrovirus) [34], foamy viruses (spumaviruses) [55], as well as the equine infectious anemia virus (EIAV, lentivirus) [3]. In addition to retroviruses, hepatitis B virus (HBV, a hepadnavirus) and adeno-associated virus (AAV, a parvovirus) are also susceptible to members of the APOBEC3 family [31, 32].
More recent work has focused on inducing HIV-1 lethal mutagenesis using drugs that have been approved for human use with other clinical indications and where cytotoxicity was likely manageable. A study with 5-azacytidine (5-AZC) demonstrated that HIV-1 lethal mutagenesis could efficiently occur at drug concentrations that had no cell cytotoxicity [19]. Since 5-AZC is a ribonucleoside analog, it was originally thought[26] that its antiviral activity would primarily be attributed to its incorporation into viral RNA and subsequent increase in HIV-1 mutation frequency [19]. In support of this, several studies have shown that 5-AZC can be incorporated into RNA [56–59]. One study further demonstrated that 5-AZC was a weak competitive inhibitor, having a 20-fold lower affinity than CTP for RNA polymerase II [60]. However, the HIV-1 lethal mutagenesis study with 5-AZC showed that the most potent antiviral activity of 5-AZC was associated with its effect on the early phase of HIV-1 replication (i.e., reverse transcription) rather than on the late phase of HIV-1 replication (i.e., RNA transcription by RNA polymerase II). In particular, while 5-AZC increased HIV-1 mutation rate in both the late and early phases of HIV-1 replication, it had the greatest effect on the early phase of replication. These data provide evidence that 5-AZC exerts its antiviral activity on both phases of replication through an increase in mutation rate. Although 5-AZC led to a modest increase in mutant frequency, similar increases in mutation rates have been shown to be sufficient to lethally mutagenize other RNA viruses [1, 8, 20, 23, 61]. As alluded to above, lethal mutagenesis modeling studies suggest that small increases in viral mutation rates should lead to a disproportionately larger decrease in viral infectivity [62, 63]. A recent experimental study with HIV-1 helps support this interrelationship between mutation rate and viral fitness [64].
A model to explain the mechanism for the 5-AZC antiretroviral effect on the early phase of the HIV-1 replication cycle was proposed. In this model, 5-AZC is first reduced by ribonucleotide reductase and is incorporated into viral DNA during reverse transcription (Figure 3). Once incorporated into DNA, the mutagenic base undergoes a spontaneous ring-opening step [65] resulting in a mispair with cytosine during synthesis of the plus strand [66]. Such a model implicates integration as a key step in repairing the induced mutations, and that the azapyrimidine base is excised by host DNA repair machinery. The incorporated mutatgenic base would then be replaced with guanosine, since it can base pair with the cytosine located in the plus-strand DNA opposite the abasic site – resulting in a G-to-C mutation. The incorporation of 5-AZC during plus strand viral DNA synthesis would ultimately result in DNA repair that would not create mutations. Therefore, HIV-1 lethal mutagenesis that is mediated by 5-AZC would specifically result in G-to-C hypermutation, which is what was observed [19, 67].
Figure 3.

A model for 5-azacytidine (5-AZC)-mediated HIV-1 mutagenesis during the minus-strand DNA synthesis step of reverse transcription. In this model, ribonucleotide reductase is proposed to convert 5-AZC-diphosphate (5-AZCDP) to 5-aza-2'-deoxycytidine diphosphate (5-aza-dCDP); 5-aza-2'-deoxycytidine (Z) triphosphate is then incorporated into the minus-strand DNA. Once incorporated, a spontaneous cytosine ring opening and deformylation of Z leads to a guanylurea derivative [65]. This allows base pairing of Z with deoxycytidine (C). Once the viral DNA is integrated into the host cell genome, Z is excised from the viral plus-strand DNA by the host cell DNA repair machinery and guanosine is incorporated into the abasic site. When transcribed into viral RNA, the guanosine in the minus-strand DNA encodes for a cytosine, thereby leading to a G-to-C transversion mutation in the plus-strand viral RNA.
Dapp et al
As APOBEC3 proteins can attack cytosine residues in minus-strand HIV-1 DNA, a recent study explored the concomitant lethal mutagenesis of both A3G and 5-AZC. Reduced viral infectivity and increased viral mutagenesis was observed with both the 5-AZC (i.e., G-to-C mutations) and A3G (i.e., G-to-A mutations); however, when combined, these mutagens had complex interactions. Nucleotide sequence analysis revealed that concomitant HIV-1 exposure to both 5-AZC and A3G resulted in an increase of G-to-A viral mutagenesis at the expense of G-to-C mutagenesis. A3G catalytic activity was required for the diminution in G-to-C mutagenesis. These studies are particularly important in providing the first indication for potentiation of the mutagenic effect of a cytosine analog by A3G expression, which can result in concomitant HIV-1 lethal mutagenesis.
Recent studies have shown that a combination of two clinically-approved cytosine analogs, gemcitabine (2'-deoxy-2',2'-difluorocytidine, a ribonucleotide reductase inhibitor) and decitabine (5-aza-2'-deoxycytidine, a cytosine analog mutagen similar to KP-1461), represent a promising new experimental approach to extinction of HIV-1 infectivity [68]. In combination, these two drugs reduced HIV-1 infectivity by 73% at concentrations that had minimal antiviral activity when used individually [68]. The observed decrease in viral infectivity coincided with a significant increase in mutation rate and resulted in a shift in the HIV-1 mutation spectrum. Analysis of HIV-1 DNA synthesis during reverse transcription indicated that there was a lack of correlation between the decitabine and gemcitabine infectivity loss and reduction in viral DNA synthesis. Therefore, these results indicated that lethal mutagenesis was the primary antiviral mechanism for inhibiting the generation of infectious progeny virus from provirus. Given that both are clinically approved drugs, there is clearly hope that clinical translation and testing of in vivo lethal mutagenesis could occur. Such studies in which drug combinations have been used to enhance HIV-1 mutagenesis have been extended to the combination of decitabine and resveratrol (a natural product known to possess inhibitory activity against ribonucleotide reductase) [69]. It should be noted that viral mutagens could perhaps be more effective in causing virus extinction when used with standard non-mutagen antivirals. For example, in tissue culture studies performed with FMDV (an RNA virus), viral populations were rapidly driven to extinction only in sequential treatment when the inhibitor was given first, followed by the viral mutagen [70]. This was far superior to the reversed sequential treatment or the simultaneous combination of inhibitor and mutagen.
Preclinical evaluation of decitabine and gemcitabine was recently reported in a mouse model for HIV-1 [71, 72]. These studies were the first to demonstrate that gemcitabine inhibits replication of a related retrovirus, murine leukemia virus (MuLV), in vivo using the MuLV-based LP-BM5/murine AIDS (MAIDS) mouse model at doses that were not toxic. Antiviral efficacy and toxicity of the drug combination in the same animal model was also investigated. The data demonstrated that the drug combination inhibited disease progression, as detected by histopathology, viral loads, and spleen weights, at doses lower than those that would be required if the drugs were used individually. The combination of decitabine and gemcitabine was found to exert antiviral activity at doses that were not toxic. Detailed analysis of provirus mutational loads was not reported, and such studies are needed to confirm that the observed antiretroviral activity is primarily associated with elevation of mutational loads. Nonetheless, such studies are promising in the establishment of in vivo models to investigate HIV-1 lethal mutagenesis as a prelude for human clinical trials, such as those conducted with KP-1461 [27].
Concluding remarks
Like many areas of AIDS research, the investigation of HIV-1 lethal mutagenesis has seen both peaks of enthusiasm and valleys of doubt. The initial enthusiasm for HIV-1 lethal mutagenesis waned once it was clear that there was no easy way to clinically translate the molecules used to induce lethal mutagenesis in cell culture. Also, concerns about the slow mutation accumulation to induce lethal mutagenesis caused apprehension over the remote possibility of virus variants emerging with new biological properties. Furthermore, concerns regarding the potential toxicity of small molecule viral mutagens must be addressed in order for the successful clinical translation of these therapeutic strategies. Such studies may include the Ames test that measures the mutagenic potential of a compound in bacteria [73], in utero studies in both rodent and non-rodent animal models to measure a chemical's teratogenic potential [74], and dose-escalation studies in animal models to assess chronic-compound exposure leading to carcinogenesis, among others (www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm065007.htm).
The discovery of the APOBEC3 proteins led to the rejuvenation of the idea of HIV-1 lethal mutagenesis as a therapeutic strategy and provided evidence of evolutionarily conserved antiviral mechanisms that are based upon lethal mutagenesis. These naturally occurring antiviral mechanisms revealed that rapid mutation accumulation within a single or limited number of rounds of replication could efficiently extinguish HIV-1 infectivity. Finally, the discovery of drugs that have the potential for clinical use have further aided in raising enthusiasm once again for HIV-1 lethal mutagenesis as an intervention strategy. In regards to clinical translation, viral mutagens may be more effective in causing virus extinction when used with standard non-mutagen antivirals. Studies with FMDV (an RNA virus) revealed rapid extinction only with sequential treatment of the inhibitor first, followed by the viral mutagen [70]. Careful studies with HIV-1 should be conducted for similar consequences. Clearly, much basic science remains to be done, but a path towards clinical translation appears more realistic than was thought in the past.
Highlights
The basis and potential cell toxicity of HIV-1 lethal mutagenesis is complicated
Important encouragement occurred from the discovery of the APOBEC3 proteins
Early thoughts of lethal mutagenesis involved the slow accumulation of mutations
Attention is now refocused on rapid mutation accumulation and low cell toxicity
Acknowledgements
This research was supported from NIH R01 GM56615, a Center for Drug Design Funding Agreement, and an AHC Translational Research Grant. M.J.D. was supported from NIH T32 DA007097.
Glossary
- Ablated mutagenesis
mutation during virus replication at lower mutation rates.
- Enhanced mutagenesis
mutation during virus replication at elevated mutation rates that do not extinguish infectivity and contribute to the genetic variability of the population.
- Error catastrophe
consensus `master' nucleotide sequence within the virus population is no longer the most fit, resulting in genetic drift in sequence space.
- Error threshold
the limit in nucleotide sequence viability at a given `critical' mutation rate, beyond which population structure breaks down and disperses over sequence space.
- Lethal mutagenesis
extinction of both the master sequence as well as all other viable viral sequences in the virus population, i.e., extinction catastrophe.
- Lethal defection
defective, replication-competent viruses that interfere with the replication of non-defective virus, resulting in overall suppression in viral replication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loeb LA, et al. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. 1999;96:1492–1497. doi: 10.1073/pnas.96.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RS, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 3.Mangeat B, et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, et al. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domingo E, et al. Quasispecies dynamics and RNA virus extinction. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Domingo E, et al. Viral quasispecies evolution. Microbiol Mol Biol Rev. 2012;76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perales C, et al. The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 2012 doi: 10.1016/j.tim.2012.08.010. In press, doi: 10.1016/j.tim.2012.1008.1010. [DOI] [PubMed] [Google Scholar]
- 8.Holland JJ, et al. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 1990;64:3960–3962. doi: 10.1128/jvi.64.8.3960-3962.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domingo E, et al. Viruses as quasispecies: biological implications. Curr Top Microbiol Immunol. 2006;299:51–82. doi: 10.1007/3-540-26397-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez-Lopez C, et al. Preextinction viral RNA can interfere with infectivity. J Virol. 2004;78:3319–3324. doi: 10.1128/JVI.78.7.3319-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grande-Perez A, et al. Suppression of viral infectivity through lethal defection. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4448–4452. doi: 10.1073/pnas.0408871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iranzo J, et al. Tempo and mode of inhibitor-mutagen antiviral therapies: a multidisciplinary approach. Proc Natl Acad Sci U S A. 2011;108:16008–16013. doi: 10.1073/pnas.1110489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manrubia SC, et al. Pathways to extinction: beyond the error threshold. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2010;365:1943–1952. doi: 10.1098/rstb.2010.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perales C, et al. Insights into RNA virus mutant spectrum and lethal mutagenesis events: replicative interference and complementation by multiple point mutants. J Mol Biol. 2007;369:985–1000. doi: 10.1016/j.jmb.2007.03.074. [DOI] [PubMed] [Google Scholar]
- 15.Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS pathogens. 2005;1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vignuzzi M, et al. Engineering attenuated virus vaccines by controlling replication fidelity. Nature Medicine. 2008;14:154–161. doi: 10.1038/nm1726. [DOI] [PubMed] [Google Scholar]
- 17.Denny WA. DNA minor groove alkylating agents. Curr Med Chem. 2001;8:533–544. doi: 10.2174/0929867003373283. [DOI] [PubMed] [Google Scholar]
- 18.Bull JJ, et al. Theory of lethal mutagenesis for viruses. Journal of Virology. 2007;81:2930–2939. doi: 10.1128/JVI.01624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dapp MJ, et al. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. Journal of virology. 2009;83:11950–11958. doi: 10.1128/JVI.01406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris KS, et al. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 2005;67:1–9. doi: 10.1016/j.antiviral.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Crotty S, et al. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 22.Crotty S, et al. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sierra S, et al. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J Virol. 2000;74:8316–8323. doi: 10.1128/jvi.74.18.8316-8323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Jarabo CM, et al. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) Virology. 2003;308:37–47. doi: 10.1016/s0042-6822(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 25.Contreras AM, et al. Viral RNA mutations are region specific and increased by ribavirin in a fulllength hepatitis C virus replication system. J Virol. 2002;76:8505–8517. doi: 10.1128/JVI.76.17.8505-8517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuevas JM, et al. The effect of ribavirin on the mutation rate and spectrum of Hepatitis C virus in vivo. J Virol. 2009;83:5760–5764. doi: 10.1128/JVI.00201-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullins JI, et al. Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PloS one. 2011;6:e15135. doi: 10.1371/journal.pone.0015135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hicks C, et al. Safety, tolerability and efficacy of KP-1461 as monotherapy for 124 days in antiretroviral-experienced, HIV-1-infected subjects. AIDS research and human retroviruses. 2012 doi: 10.1089/aid.2012.0093. doi:10.1089/aid.2012.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brok J, et al. Ribavirin plus interferon versus interferon for chronic hepatitis C. Cochrane database of systematic reviews. 2010:CD005445. doi: 10.1002/14651858.CD005445.pub2. [DOI] [PubMed] [Google Scholar]
- 30.Lecossier D, et al. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 31.Turelli P, et al. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, et al. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Current biology : CB. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 33.Bishop KN, et al. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Current biology : CB. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 34.Sasada A, et al. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology. 2005;2:32. doi: 10.1186/1742-4690-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariani R, et al. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 36.Zennou V, Bieniasz PD. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology. 2006;349:31–40. doi: 10.1016/j.virol.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 38.Malim MH, Emerman M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell host & microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annual review of immunology. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 40.Browne EP, et al. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology. 2009;387:313–321. doi: 10.1016/j.virol.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shindo K, et al. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. The Journal of biological chemistry. 2003;278:44412–44416. doi: 10.1074/jbc.C300376200. [DOI] [PubMed] [Google Scholar]
- 42.Bishop KN, et al. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schumacher AJ, et al. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. Journal of virology. 2008;82:2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armitage AE, et al. APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete “all or nothing” phenomenon. PLoS genetics. 2012;8:e1002550. doi: 10.1371/journal.pgen.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chelico L, et al. Stochastic properties of processive cytidine DNA deaminases AID and APOBEC3G. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2009;364:583–593. doi: 10.1098/rstb.2008.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadler HA, et al. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. Journal of virology. 2010;84:7396–7404. doi: 10.1128/JVI.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jern P, et al. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS pathogens. 2009;5:e1000367. doi: 10.1371/journal.ppat.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim EY, et al. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. Journal of virology. 2010;84:10402–10405. doi: 10.1128/JVI.01223-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulder LC, et al. Cytidine deamination induced HIV-1 drug resistance. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5501–5506. doi: 10.1073/pnas.0710190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebrahimi D, et al. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. Journal of virology. 2011;85:9139–9146. doi: 10.1128/JVI.00658-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pace C, et al. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. Journal of virology. 2006;80:9259–9269. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez-Perez JA, et al. APOBEC3G mRNA expression in exposed seronegative and early stage HIV infected individuals decreases with removal of exposure and with disease progression. Retrovirology. 2009;6:23. doi: 10.1186/1742-4690-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albin JS, Harris RS. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert reviews in molecular medicine. 2010;12:e4. doi: 10.1017/S1462399409001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenberg BR, Papavasiliou FN. Beyond SHM and CSR: AID and related cytidine deaminases in the host response to viral infection. Advances in immunology. 2007;94:215–244. doi: 10.1016/S0065-2776(06)94007-3. [DOI] [PubMed] [Google Scholar]
- 55.Delebecque F, et al. Restriction of foamy viruses by APOBEC cytidine deaminases. Journal of virology. 2006;80:605–614. doi: 10.1128/JVI.80.2.605-614.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paces V, et al. Incorporation of 5-azacytidine into nucleic acids of Escherichia coli. Biochimica et biophysica acta. 1968;161:352–360. [PubMed] [Google Scholar]
- 57.Li LH, et al. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer research. 1970;30:2760–2769. [PubMed] [Google Scholar]
- 58.Doskocil J, Sorm F. The action of 5-azacytidine on bacteria infected with bacteriophage T4. Biochim Biophys Acta. 1967;145:780–791. [PubMed] [Google Scholar]
- 59.Cihak A, et al. Incorporation of 5-azacytidine into liver ribonucleic acids of leukemic mice sensitive and resistant to 5-azacytidine. Biochim Biophys Acta. 1965;108:516–518. doi: 10.1016/0005-2787(65)90046-8. [DOI] [PubMed] [Google Scholar]
- 60.Lee TT, Momparler RL. Kinetic studies with 5-azacytidine-5'-triphosphate and DNA-dependent RNA polymerase. Biochem Pharmacol. 1977;26:403–406. doi: 10.1016/0006-2952(77)90199-x. [DOI] [PubMed] [Google Scholar]
- 61.Julias JG, Pathak VK. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 1998;72:7941–7949. doi: 10.1128/jvi.72.10.7941-7949.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eigen M. Error catastrophe and antiviral strategy. Proc Natl Acad Sci U S A. 2002;99:13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Domingo E. Quasispecies and the development of new antiviral strategies. Prog Drug Res. 2003;60:133–158. doi: 10.1007/978-3-0348-8012-1_4. [DOI] [PubMed] [Google Scholar]
- 64.Dapp MJ, et al. Interrelationship between HIV-1 fitness and mutation rate. Journal of Molecular Biology. 2012 doi: 10.1016/j.jmb.2012.10.009. In press, DOI: 10.1016/j.jmb.2012.1010.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogstad DK, et al. Chemical decomposition of 5-aza-2'-deoxycytidine (Decitabine): kinetic analyses and identification of products by NMR, HPLC, and mass spectrometry. Chemical research in toxicology. 2009;22:1194–1204. doi: 10.1021/tx900131u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jackson-Grusby L, et al. Mutagenicity of 5-aza-2'-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc Natl Acad Sci U S A. 1997;94:4681–4685. doi: 10.1073/pnas.94.9.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dapp MJ, et al. Concomitant lethal mutagenesis of human immunodeficiency virus type 1. Journal of molecular biology. 2012;419:158–170. doi: 10.1016/j.jmb.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clouser CL, et al. Exploiting drug repositioning for discovery of a novel HIV combination therapy. Journal of virology. 2010;84:9301–9309. doi: 10.1128/JVI.01006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clouser CL, et al. Anti-HIV-1 activity of resveratrol derivatives and synergistic inhibition of HIV-1 by the combination of resveratrol and decitabine. Bioorg Med Chem Lett. 2012 doi: 10.1016/j.bmcl.2012.08.108. doi: 10.1016/j.bmcl.2012.1008.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perales C, et al. Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS pathogens. 2009;5:e1000658. doi: 10.1371/journal.ppat.1000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clouser CL, et al. Analysis of the ex vivo and in vivo antiretroviral activity of gemcitabine. PLoS One. 2011;6:e15840. doi: 10.1371/journal.pone.0015840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clouser CL, et al. Activity of a novel combined antiretroviral therapy of gemcitabine and decitabine in a mouse model for HIV-1. Antimicrob Agents Chemother. 2012;56:1942–1948. doi: 10.1128/AAC.06161-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ames BN, et al. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Natl Acad Sci U S A. 1973;70:782–786. doi: 10.1073/pnas.70.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ito T, et al. Teratogenic effects of thalidomide: molecular mechanisms. Cellular and molecular life sciences : CMLS. 2011;68:1569–1579. doi: 10.1007/s00018-010-0619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]