

Abstract
Context:
Isolated hypogonadotropic hypogonadism (IHH) is caused by defective GnRH secretion or action resulting in absent or incomplete pubertal development and infertility. Most women with IHH ovulate with physiological GnRH replacement, implicating GnRH deficiency as the etiology. However, a subset does not respond normally, suggesting the presence of defects at the pituitary or ovary.
Objectives:
The objective of the study was to unmask pituitary or ovarian defects in IHH women using a physiological regimen of GnRH replacement, relating these responses to genes known to cause IHH.
Design, Setting, and Subjects:
This study is a retrospective analysis of 37 IHH women treated with iv pulsatile GnRH (75 ng/kg per bolus).
Main Outcome Measures:
Serum gonadotropin and sex steroid levels were measured, and 14 genes implicated in IHH were sequenced.
Results:
During their first cycle of GnRH replacement, normal cycles were recreated in 60% (22 of 37) of IHH women. Thirty percent of women (12 of 37) demonstrated an attenuated gonadotropin response, indicating pituitary resistance, and 10% (3 of 37) exhibited an exaggerated FSH response, consistent with ovarian resistance. Mutations in CHD7, FGFR1, KAL1, TAC3, and TACR3 were documented in IHH women with normal cycles, whereas mutations were identified in GNRHR, PROKR2, and FGFR1 in those with pituitary resistance. Women with ovarian resistance were mutation negative.
Conclusions:
Although physiological replacement with GnRH recreates normal menstrual cycle dynamics in most IHH women, hypogonadotropic responses in the first week of treatment identify a subset of women with pituitary dysfunction, only some of whom have mutations in GNRHR. IHH women with hypergonadotropic responses to GnRH replacement, consistent with an additional ovarian defect, did not have mutations in genes known to cause IHH, similar to our findings in a subset of IHH men with evidence of an additional testicular defect.
Isolated hypogonadotropic hypogonadism (IHH) is a rare genetic disorder that results in failure to undergo normal pubertal development. Patients with IHH harbor mutations in genes involved in GnRH neuronal migration as well as GnRH synthesis and secretion (1). In most patients with IHH, physiological GnRH replacement restores normal levels of pituitary and gonadal hormones, allowing for testicular growth and spermatogenesis in men and ovulation in women (2, 3), thus implicating GnRH deficiency as the primary underlying etiology of the reproductive phenotype in IHH patients.
However, decreased pituitary responsiveness to GnRH has been described in IHH women in association with mutations in the GnRH receptor (4), indicating that in these patients, the defect lies at the level of the pituitary. Furthermore, GnRH replacement studies have unmasked functional defects at the level of the testes as well as the pituitary in subsets of IHH men (3). These findings are of particular interest because our growing understanding of the genetic architecture of IHH indicates that many of the genes that cause this syndrome are expressed in the mammalian pituitary (5–10) or gonad (11–19) in addition to the brain.
We therefore hypothesized that identification of women with abnormal responses to physiological GnRH replacement would provide greater insight into the pathogenesis of IHH. Hypo- or hypergonadotropic responses to GnRH would implicate pituitary or ovarian defects, respectively. Such findings would suggest that genes involved in gonadotrope or ovarian development and function that are also expressed in the hypothalamus should be given greater consideration in the search for new IHH genes.
Materials and Methods
IHH subjects
Thirty-seven women with IHH who received iv pulsatile GnRH treatment at a dose of 75 ng/kg per bolus during their first treatment cycle at the Reproductive Endocrine Unit of the Massachusetts General Hospital between 1980 and 2012 were included in this study. An additional 63-year-old IHH woman was treated with iv GnRH for 7 days to provide a basis for comparison of FSH levels in younger subjects found to have an ovarian defect. The diagnostic criteria for IHH have been previously described in detail (20). This study was approved by the Partners Human Research Committee and signed informed consent was obtained from each subject prior to participation.
Baseline assessment
A detailed history was taken and physical examination performed on all subjects. Obesity was defined as a body mass index (BMI) greater than 30 kg/m2. Subjects were classified as anosmic based on a 40-item University of Pennsylvania Smell Identification Test score of the fifth percentile or less for age (21) or on self-report. Hormone replacement therapy was discontinued for at least 1 month prior to the baseline biochemical assessment. Frequent blood sampling (every 10 minutes for 12 hours) was performed in all subjects to assess endogenous GnRH secretion as manifested by LH pulsatility. FSH, estradiol (E2), and progesterone (P4) were assayed from pools of the frequent sampling studies. Pulsatile LH secretion was analyzed as previously described (22, 23), and studies were categorized as pulsatile or apulsatile.
Response to physiological GnRH replacement
Pulsatile GnRH was administered using a closed iv system, as previously described (2). Daily blood samples for LH, FSH, E2, and P4 were drawn 40 minutes after a GnRH bolus at the same time of day. Beginning on day 7, serial pelvic ultrasounds were performed to monitor follicular growth and document ovulation. Women with IHH who did not ovulate in response to the standard dose of GnRH received additional treatment at the same or higher doses.
Genotyping
Genomic DNA was obtained from peripheral blood lymphocytes in 31 of 37 subjects. Exonic and proximal intronic (>15 bp from splice sites) DNA sequences of 14 genes implicated in the etiology of IHH were amplified by PCR and determined by direct sequencing. These genes include KAL1 (anosmin-1), GNRH1 (GnRH 1), GNRHR (GnRH receptor), KISS1 (kisspeptin), KISS1R (KISS1 receptor), NELF (nasal embryonic LHRH factor), FGF8 (fibroblast growth factor 8), FGFR1 (fibroblast growth factor receptor 1), PROK2 (prokineticin 2), PROKR2 (prokineticin receptor 2), TAC3 (tachykinin 3), TACR3 (tachykinin receptor 3), DAX1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1), and CHD7 (chromodomain-helicase-DNA binding protein 7). PCR primers and amplification conditions for each gene have been described previously (24–33). Mutations were defined as rare sequence variants (RSVs) that were: 1) within coding regions ± 5 bp; 2) nonsynonymous; 3) present in less than 1% of alleles in 192 unaffected controls who had normal reproductive function by history and physical examination; and 4) not categorized as polymorphisms by the 1000 Genomes Browser (http://browser.1000genomes.org/).
Functional analyses
The in vitro functional data for the FGFR1, GNRHR, PROK2, and PROKR2 genes have been previously reported (25, 26, 32, 34–37). In instances in which in vitro data were not available, 5 different prediction algorithms were used to determine the potential significance of missense variants and 1 for intronic changes [PolyPhen (38), Mutation Taster (39), Panther (40), SIFT (41), pMUT (42), and Human Splicing Finder (43)].
Controls
One hundred three unaffected, regularly cycling (25–35 days) women, aged 19–42 years, provided daily blood samples across a complete menstrual cycle. All cycles were determined to be ovulatory based on a luteal phase P4 level of greater than 6 ng/mL (19 nmol/L). Eighty-one of these women served as controls in a previous study (44).
Assays
Serum LH and FSH were analyzed by RIA or using a 2-site monoclonal nonisotopic system (Axsym; Abbott Laboratories, Abbott Park, Illinois) calibrated with the same reference preparation used in the RIA (45) and expressed in international units per liter of the Pituitary Second International Standard 80/552. E2 was initially measured by RIA and then by using a 2-site monoclonal nonisotopic system (Axsym; Abbott Laboratories) with a functional sensitivity of 20 pg/mL (73.4 pmol/L), as previously described (46). P4 was measured by RIA or using a chemiluminescent enzyme immunoassay (ELISA; Immulite, Diagnostic Products Corp, Los Angeles, California) as previously described (46).
Data analysis
In subjects and controls, the day of ovulation was defined according to previously validated criteria (47). Data from the daily sampling protocol in controls were used to interpret the gonadotropin responses to GnRH observed in IHH women. Ovulation occurred no later than cycle day 20 in the controls. Therefore, GnRH treatment cycles were classified as normal if ovulation occurred on or prior to treatment day 20 and abnormal if ovulation occurred after day 20 or if the subject did not ovulate.
Gonadotropin and E2 levels during cycle days 1–7 in controls and during the first 7 days of pulsatile GnRH replacement (75 ng/kg per bolus) in IHH women with normal cycles were expressed as interquartile ranges [(IQRs) 25th to the 75th centile]. Hormonal data from IHH women with normal cycles were referenced against that from controls. Data from IHH women with abnormal responses to GnRH were then referenced against IHH women with normal cycles. Women with abnormal responses demonstrated either an inadequate gonadotropin response to GnRH consistent with pituitary resistance or an exaggerated FSH response to GnRH, indicating underlying ovarian resistance.
Phenotypic and genotypic characteristics of women in each group were compared using ANOVA and expressed as mean ± SD or using χ2. BMI was not normally distributed and was expressed as median and IQR. Linear regression was used to determine the relationship between age and peak FSH level during the first 7 days of treatment in IHH women with normal cycles. Peak FSH was log transformed prior to analysis. A similar analysis was performed to determine whether there was an inverse relationship between BMI and peak LH in IHH women with normal cycles as has been demonstrated in both normal cycling women (48) and in women with polycystic ovarian syndrome (48–50). P < .05 was considered to be significant unless otherwise noted.
Results
Baseline characteristics
The clinical and biochemical features of the 37 women with IHH are summarized in Tables 1 and 2. All subjects were 16 years old or older at the time of treatment (mean age 29.8 ± 7.3 years), had low E2 levels in the face of low or inappropriately normal gonadotropins, no other pituitary hormone deficiencies, and no neuroanatomic cause of IHH. Forty-six percent of the women were anosmic.
Table 1.
Baseline Characteristics of the Cohort of Women With IHH Treated With iv GnRH
| Normal IHH | Abnormal IHH |
||
|---|---|---|---|
| Pituitary Resistance | Ovarian Resistance | ||
| n | 22 | 12 | 3 |
| Anosmia, % | 55% | 42% | 0%a,b |
| Age, y | 28.5 ± 7.4 (16.8–41.9) | 30.5 ± 6.9 (17.6–39.4) | 36.0 ± 4.9a (31.2–41.0) |
| BMI, kg/m2, median (IQR) | 22.4 (19.2–25.9) | 28.0 (22.9–36.5)b | 22.8 (22.6–25.9) |
| Partial puberty, % | 46% | 75% | 0% |
| FSH, IU/L | 0.87 (0.24–2.87) | 0.44 (0.22–0.99)b | 0.57 (0.50–0.70)b |
| LH, IU/L | 0.28 (0.09–1.00) | 0.20 (0.08–0.44) | 0.34 (0.23–1.19) |
| Apulsatile baseline study, % | 52% | 83% | 100%b |
| Ovarian volume, mLc | 1.6 (1.4–2.7) | 1.9 (1.0–2.2) | 1.5 (1.1–2.4) |
P < .05 vs pituitary resistance.
P < .05 vs normal response cycles.
Mean ovarian volume in normal spontaneously cycling women is 9.5 mL (95% confidence interval 3.9–15.9 mL) (48).
Table 2.
Phenotypic and Genotypic Characteristics of Women With IHH Treated With iv GnRH
| Subject no. | Age, y | BMI, kg/m2 | Baseline Study | Partial Puberty | Olfactory Phenotype | Nonreproductive Phenotype | Gonadotropin Levels (d 1–7)a |
Genetics |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FSH | LH | Gene | Variant | Functional Data (Reference) | Prediction Programsb | |||||||
| Normal response | ||||||||||||
| 1 | 20.0 | 22.7 | A | N | nIHH | High arched palate, scoliosis | TACR3 | p.W208X | Nonsense | |||
| 2 | 22.8 | 21.3 | A | N | nIHH | Neg | ||||||
| 3 | 36.9 | 23.5 | P | Y | nIHH | Cleft lip and palate, widely-spaced teeth | Neg | |||||
| 4 | 28.9 | 20.0 | A | N | nIHH | Partial empty sella | N.A. | |||||
| 5 | 36.4 | 28.3 | P | Y | nIHH | Marfanoid habitus | Neg | |||||
| 6 | 17.6 | 18.0 | A | N | KS | Ocular defect | CHD7 | c.3202–5 T>C (splice) | 0c | |||
| 7 | 26.6 | 33.6 | A | Y | KS | Ocular defect, short fourth metacarpal | FGFR1 | p.L630P | 4 | |||
| 8 | 33.2 | 18.0 | A | N | KS | Neg | ||||||
| 9 | 38.6 | 25.4 | P | Y | KS | Hearing loss | KAL1 | p.K185N | 2 | |||
| 10 | 31.5 | 18.8 | A | N | KS | Neg | ||||||
| 11 | 23.0 | 21.7 | P | N | KS | N.A. | ||||||
| 12 | 16.8 | 21.5 | P | N | KS | Neg | ||||||
| 13 | 18.5 | 18.9 | A | N | KS | Clinodactyly | Neg | |||||
| 14 | 31.8 | 25.9 | P | Y | KS | Neg | ||||||
| 15 | 25.5 | 24.3 | P | Y | nIHH | N.A. | ||||||
| 16 | 41.9 | 35.6 | P | Y | nIHH | Neg | ||||||
| 17 | 28.5 | 22.1 | P | N | nIHH | Neg | ||||||
| 18 | 23.8 | 25.6 | A | N | nIHH | TAC3 | p.R80S | 4 | ||||
| 19 | 38.6 | 18.2 | A | N | KS | Flat nasal bridge, PDA, hypoplastic fifth finger (CHAR syndrome) | N.A. | |||||
| 20 | 32.2 | 26.5 | P | Y | KS | Neg | ||||||
| 21 | 34.8 | 18.1 | A | Y | nIHH | N.A. | ||||||
| 22 | 19.9 | 26.9 | A | Y | KS | Widely-spaced teeth | CHD7 | p.L373F | 1 | |||
| Abnormal, pituitary resistance | ||||||||||||
| 23 | 23.4 | 20.9 | A | Y | nIHH | ↓ | ↓ | Neg | ||||
| 24 | 27.9 | 21.7 | P | N | KS | Short fourth metacarpal | NI | ↓ | FGFR1, GNRHR | p.R470L; p.Q106R, p.R262 (all partiald) | (26, 35, 37) | |
| 25 | 39.4 | 42.8 | A | Y | KS | Clinodactyly, flat nasal bridge | ↓ | ↓ | Neg | |||
| 26 | 30.0 | 23.3 | A | N | nIHH | Partial empty sella | NI | ↓ | PROKR2 | p.L173R (partial) | (25) | |
| 27 | 35.5 | 28.2 | A | Y | nIHH | ↓ | ↓ | GNRHR | p.M1T + p.R139H (complete); p.Q106R (partial) | Start codon (35) | ||
| 28 | 29.9 | 43.8 | A | Y | nIHH | ↓ | ↓ | Neg | ||||
| 29 | 43.0 | 36.0 | A | Y | nIHH | Widely-spaced teeth | ↓ | ↓ | Neg | |||
| 30 | 27.1 | 37.7 | A | Y | KS | NI | ↓ | Neg | ||||
| 31 | 33.5 | 27.7 | A | Y | nIHH | ↓ | ↓ | N.A. | ||||
| 32 | 17.6 | 26.4 | A | N | KS | ↓ | ↓ | Neg | ||||
| 33 | 32.3 | 32.4 | A | Y | KS | Syndactyly, pes planus | ↓ | ↓ | Neg | |||
| 34 | 26.7 | 20.1 | P | Y | nIHH | NI | ↓ | GNRHR | p.N10K+p.Q11K (partial); p.P320L (complete) | (34, 36) | ||
| Abnormal, ovarian resistance | ||||||||||||
| 35 | 41.0 | 22.3 | A | N | nIHH | ↑ | ↑ | Neg | ||||
| 36 | 35.9 | 22.8 | A | N | nIHH | Mitral valve prolapse | ↑ | ↑ | Neg | |||
| 37 | 31.2 | 28.9 | A | N | nIHH | Cleft palate | ↑ | NI | Neg | |||
| 63-year-old woman | ||||||||||||
| 38 | 63.0 | 23.5 | A | N | nIHH | ↑ | ↑ | PROK2 | p.155fsX1 (complete) | (32) | ||
Abbreviations: A, apulsatile; KS, Kallmann's (anosmic or hyposmic IHH); N, no; N.A., DNA sample not available; Neg, negative; NI, normal; nIHH, normosmic IHH; P, pulsatile; Y, yes; ↑, high; ↓, low.
Gonadotropin levels during the first week of GnRH treatment relative to normal IHH responders.
Number of prediction programs (of 5) predicting this mutation to be deleterious.
A single prediction program, Human Splicing Finder, does not predict that this variant will alter splicing.
Refers to partial or complete loss of function.
Characterization of response to physiological pulsatile GnRH replacement
Normal response (n = 22)
Sixty percent of IHH women (22 of 37) ovulated during their first cycle of pulsatile GnRH at a dose of 75 ng/kg per bolus on or before day 20 (day 14.8 ± 2.2; range 11–18 days). FSH rose to the range seen in controls during the early follicular phase within 24–48 hours of GnRH treatment, whereas LH and E2 lagged slightly, reaching the normal range at 72 hours (Fig. 1). Seventy-one percent of women developed a dominant follicle (≥11 mm) by cycle day 10, in line with the follicular growth rate seen during normal menstrual cycles (51). The peak FSH response to GnRH treatment during week 1 increased with age (P = .03, r = 0.5). Baseline LH was inversely associated with BMI (P = .05, r = 0.4), but there was no relationship between BMI and baseline FSH (P = .1), peak LH (P = .2), or peak FSH (P = .9).
Figure 1.
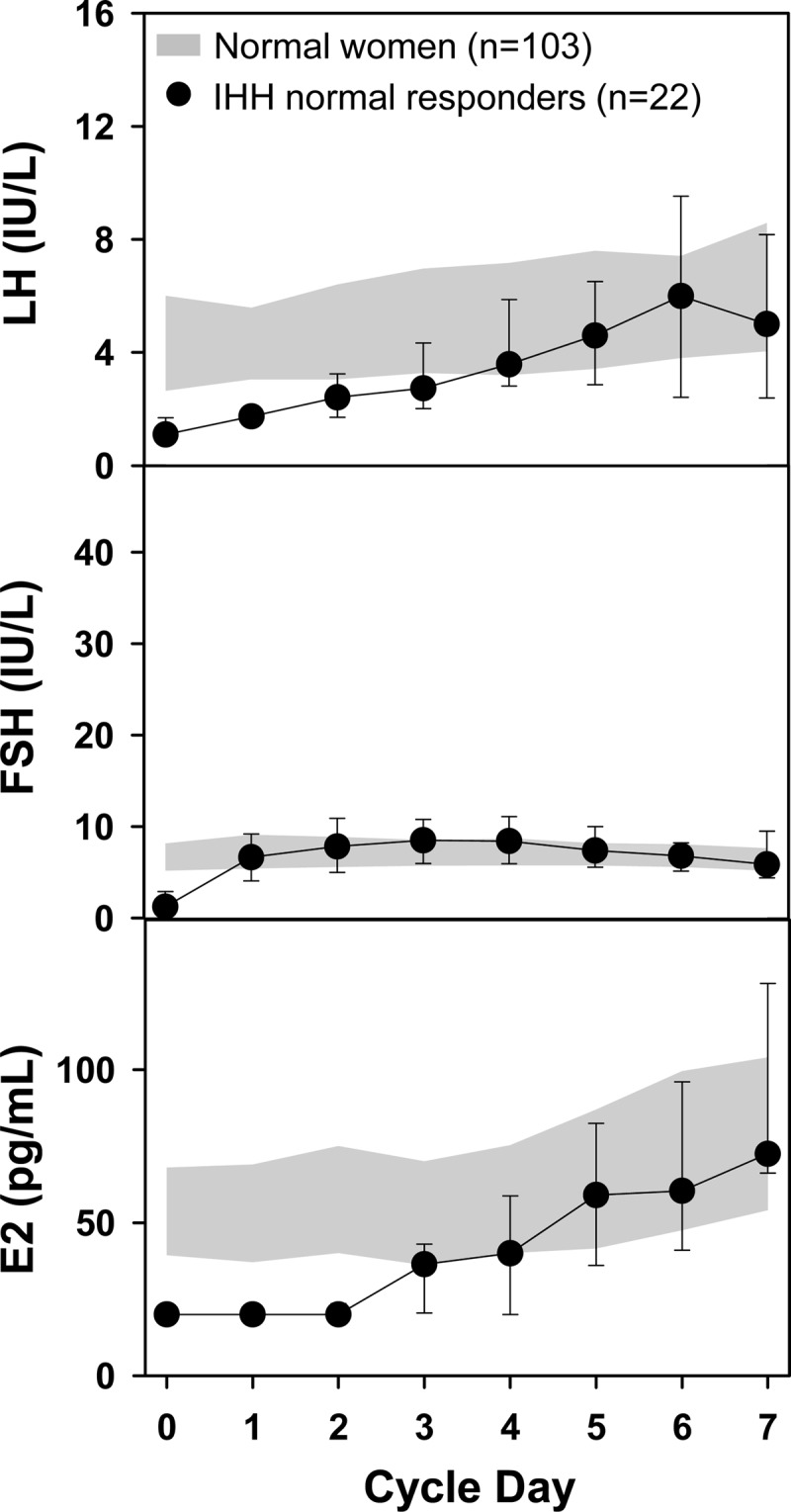
In the 22 IHH women who ovulated within the normal time frame in response to initial exposure to a physiological dose of pulsatile GnRH (75 ng/kg, iv), gonadotropin and estradiol levels (mean ± 1 SD) rose to the normal range observed in the early follicular phase of regularly cycling women (shaded area, IQR) within the first few days of treatment.
Abnormal response (n = 15)
Fifteen IHH women did not ovulate during the first 20 days of their first GnRH treatment cycle (Fig. 2).
Figure 2.

A, Gonadotropin and estradiol levels (mean ± 1 SD) remained low in response to iv pulsatile GnRH (75 ng/kg) in 12 women, suggesting resistance at the level of the pituitary. B, A hypergonadotropic response (mean ± 1 SD) was observed in 3 other women who did not ovulate during the first 20 days of the GnRH treatment cycle. The FSH response in this group is similar to that seen during the first week of GnRH therapy in a 63-year-old IHH woman (squares). Shaded region represents the interquartile range for the 22 IHH women with normal responses to GnRH.
Pituitary resistance (n = 12)
Low LH and FSH levels (n = 8).
During the first week of treatment, 8 women demonstrated defects in both FSH and LH secretion in response to GnRH relative to IHH women with normal cycles (Fig. 2A). In 5 of the 8 women, gonadotropins eventually rose into the normal range after an additional week of treatment or with an increase in dose (100–150 ng/kg), allowing for normal dominant follicle development, ovarian E2 synthesis, and ovulation (in 4 of 5). In the remaining 3 women, prolonged treatment and/or dose increases to 100 ng/kg were unable to normalize gonadotropin levels. One of the 3 chose to undergo an additional cycle with high-dose (250 ng/kg) GnRH and successfully ovulated.
Preserved FSH secretion (n = 4).
Four women demonstrated an isolated defect in LH secretion during the first 7 days of treatment. Three of the 4 grew a dominant follicle (21–30 mm); however, none ovulated until receiving higher doses of GnRH (100–150 ng/kg) in subsequent treatment cycles.
Ovarian resistance (n = 3)
Three women with IHH demonstrated an exaggerated FSH response to GnRH during the first week of treatment, with FSH levels rising significantly above the 75th centile of levels observed in IHH women with normal cycles (ages 17–42 years) or controls (age 20–42 years) (Fig. 2B). The pattern of FSH during the first 4 days of treatment in these patients closely matched the response in a 63-year-old IHH woman treated with pulsatile GnRH (Fig. 2B). All 3 women grew a dominant follicle in association with elevated FSH levels. Subject no. 37 (aged 31 years) had delayed folliculogenesis but ovulated with prolonged GnRH treatment (75 ng/kg). Subject no. 36 (aged 36 years) had persistently low E2 levels, even in subsequent cycles with higher doses of GnRH, but eventually ovulated with high-dose gonadotropin treatment (450 IU Menopur). Subject no. 35 (aged 41 years) had an LH surge but an insufficient luteal phase [peak P4 <3.0 ng/dL (9.6 pmol/L)] in response to the 75-ng/kg replacement dose. Investigation into other causes of ovarian resistance in these women revealed normal karyotypes (n = 3), negative ovarian antibodies (n = 1), and the absence of autoimmune disease (n = 3). FMR1 premutation testing was not performed.
Comparison of baseline characteristics among different response groups
There was no difference in the age of presentation among the different response groups. BMI was highest among women with abnormal/pituitary resistant responses (42% obese) (P < .04) (Table 1). Nonreproductive phenotypes previously associated with IHH were present in all groups (Table 2). Women with partial puberty (spontaneous breast development) were no more likely to demonstrate a normal response to GnRH than women with absent puberty (Table 1).
Baseline FSH, but not LH, levels were higher in women with normal responses to GnRH compared with those with abnormal responses (P < .05). E2 levels were at the limit of detection of the assay and ovarian volume was diminished in all IHH groups compared with reproductive-aged women (48) (Table 1). The higher FSH levels observed at baseline in the IHH women with normal responses is consistent with the presence of pulsatile LH secretion in nearly half of these women as slow frequency GnRH stimulation favors FSH secretion (52). The higher incidence of apulsatile LH profiles in the women with abnormal responses to GnRH may reflect a more profound defect in GnRH secretion or pituitary responsiveness to GnRH. Anosmia was present in 46% of the cohort but was limited to women with normal or pituitary-resistant response cycles (Table 1).
Comparison of genotypes among different response groups
Genotyping was performed in 17 of 22 IHH women with normal cycles and in 14 of 15 IHH women with abnormal cycles. Six women with normal cycles had a monogenic, monoallelic variant in 1 of the 14 genes examined (Table 2). There were 4 missense RSVs (FGFR1, CHD7, TAC3, and KAL1) predicted to be deleterious by 1–4 prediction programs, 1 nonsense RSV in TACR3, and 1 splice-site RSV in CHD7 not predicted to disturb splicing by the 1 prediction program used. Among the IHH women with pituitary resistance, 1 had a monoallelic loss of function (LOF) mutation in PROKR2 (25), 2 harbored 3 LOF mutations in GNRHR each (34–36), and 1 had a biallelic LOF mutation in GNRHR (26, 35) combined with a monoallelic LOF mutation in FGFR1 (53). No RSVs were identified in the women with ovarian resistance. The 63-year-old woman with IHH had a homozygous deletion in PROK2, resulting in a nonfunctional, truncated protein (32).
Discussion
In most women with IHH, physiological replacement of GnRH activates the dormant reproductive axis, allowing women who have never before been exposed to GnRH to achieve normal follicular phase levels of gonadotropins and sex steroids in a matter of days and to ovulate within 2 weeks. This finding stands in contrast to men with IHH who typically require months to a year of GnRH therapy due to the much longer time required for testicular growth and spermatogenesis (54). The rapid response to GnRH replacement in IHH women not only has immediate fertility benefits but also provides an early opportunity to identify women with abnormal responses to physiological GnRH replacement. Demonstration of hypo- or hypergonadotropic responses to GnRH localizes defects to the pituitary or ovary, respectively, that may not be apparent in the absence of GnRH.
Pituitary and testicular, in addition to hypothalamic, defects were identified by Sykiotis et al (3) in a subset of IHH men (23 of 90 men) through detailed examination of gonadotropin, testosterone, and spermatogenic responses to long-term GnRH administration. Genotype-phenotype (ie, response to GnRH) correlation studies in this group of men were limited because the atypical responders to GnRH were predominantly mutation negative (75%) or had an isolated defect in KAL1 (25%). Furthermore, it is impossible to attribute gonadal defects in adult men with IHH entirely to genetic defects due to the potential for confounding by neonatal cryptorchidism, which influences gonadal function in the adult male (55). Systematic investigation of the response to GnRH in IHH women allowed us to confirm the presence of gonadal defects in patients with IHH. In part due to the larger number of genes (14 vs 8) sequenced, we were also able to relate phenotype to genotype in the small number of IHH women with abnormal responses to GnRH who demonstrated a greater variety of genetic defects than observed in our cohort of IHH men (3).
Most women with abnormal responses to initial exposure to pulsatile GnRH had low gonadotropin levels, indicating pituitary resistance. None of these women had signs of other pituitary hormone deficiencies, and only 1 had an abnormal pituitary on magnetic resonance imaging (partial empty sella). As expected, this group encompassed all women with mutations in GNRHR. Two women with mutations in GNRHR had preserved FSH but impaired LH secretion in response to GnRH replacement (4, 36), consistent with in vitro studies demonstrating that LHβ gene transcription is more sensitive to GnRH than FSHβ (36). The third woman with a mutation in GNRHR demonstrated a profound defect in both LH and FSH responsiveness (56), potentially due to a more severe genetic defect in GnRH receptor signaling, as suggested by in vitro studies (26, 34–36). Importantly, in all women with GNRHR mutation, impaired receptor signaling was overcome with higher doses of GnRH and ovulation was achieved. Our observations in IHH women with mutations in GNRHR differ from those of Sykiotis et al (3), who observed a normal response to GnRH in an IHH man with mutations in both GNRHR and FGFR1. The differential response between patients with GNRHR mutations in the 2 studies may well be explained by differences in genetic burden; the male patient had a monoallelic partial LOF mutation in GNRHR, whereas the women in the current study had biallelic partial or biallelic partial and complete LOF mutations in GNRHR (56).
One woman with pituitary resistance to pulsatile GnRH treatment harbored a heterozygous mutation in PROKR2 (L163R), previously shown to severely compromise PROKR2 signaling and expression in vitro (25), and had a partial empty sella on magnetic resonance imaging. These findings are consistent with studies implicating PROK2 and PROKR2 in pituitary gland development. PROKR2 is expressed in the human pituitary gland (8), and patients with hypopituitarism and abnormal pituitary anatomy (57, 58) have been shown to have mutations in PROK2 and PROKR2. The patient in the current study had an isolated defect in LH secretion and achieved ovulation with a higher dose of GnRH, suggesting that pituitary resistance associated with a prokineticin signaling defect is relative and, importantly for therapy, surmountable with increased GnRH dosage. The normal follicular growth rate, E2 levels, and successful ovulation in this patient after normalization of gonadotropin levels with a higher dose of GnRH also indicate the absence of an additional ovarian defect, consistent with the observation that PROK2-deficient female mice ovulate in response to gonadotropins (32). Interestingly, an IHH man in the cohort of Sykiotis et al (3) with a heterozygous PROKR2 mutation (R164Q) that also causes a severe disruption in PROKR2 signaling and expression nearly identical to that of L163R in head-to-head studies (25), responded normally to the standard male GnRH treatment regimen. The 63-year-old IHH woman in the current series with a homozygous deletion in PROK2 (32) also responded normally to GnRH with the hypergonadotropic response expected for her age. Thus, the functional role of the prokineticin pathway at the pituitary is incompletely understood, and it is possible that a second mutation in a gene relevant to pituitary function will be identified in the pituitary-resistant IHH woman with the PROKR2 mutation that can explain her abnormal response to GnRH.
Studies in a number of animal models have demonstrated that the fibroblast growth factor family also plays an important role in early pituitary gland development (5, 6). In situ hybridization studies in human embryos localizing FGFR1 to Rathke's pouch (9), the precursor of the anterior pituitary, and the observation that patients with abnormal pituitary anatomy or function have mutations in FGF8 or FGFR1 (9, 57) strongly suggest that fibroblast growth factor signaling plays a key role in pituitary development in humans. Interestingly, the 1 IHH woman with a monogenic RSV in FGFR1 (L630P) demonstrated a normal response to GnRH, suggesting that partial defects in FGFR1 signaling alone do not interfere with GnRH action at the pituitary. Although in silico prediction programs suggest that the L630P mutation is deleterious, its effects on receptor signaling have not been investigated in vitro. Therefore, we cannot exclude the possibility that this woman demonstrated a normal response to GnRH because this RSV has a minor effect on FGFR1 function.
CHD7 is also expressed in the human (10) and mouse (59) pituitary, yet the 2 women with heterozygous mutations in CHD7 responded normally to GnRH replacement. In the absence of an in vitro assay for CHD7, it is difficult to assess the functional impact of these RSVs; however, several prediction programs suggest that they do not significantly alter protein function. Thus, these women may have been able to respond normally to GnRH because CHD7 function is preserved, and a different mutation in an unidentified gene is responsible for their IHH. Alternatively, the normal pituitary response to GnRH observed in women with mutations in CHD7, in combination with reports that mice with heterozygous loss of CHD7 demonstrate normal gonadotropin responses to a GnRH agonist (60), may indicate that the CHD7 protein acts predominantly upstream of the pituitary in hypothalamic neurogenesis and/or GnRH neuronal migration.
Importantly, two thirds of the IHH women with pituitary resistance were mutation negative, providing further evidence that additional genes important for pituitary function will be identified in the IHH patient population. Studies of transcripts coexpressed in the pituitary and hypothalamus suggest that mutations in genes involved in cell differentiation, intracellular transduction, vesicle transport, or transcription may yet be found in patients with IHH (61).
Overall, IHH women with pituitary resistance tended to be heavier than the other groups. An inverse relationship between BMI and LH responsiveness to GnRH has previously been reported in regularly cycling women and women with polycystic ovarian syndrome (48–50), and in the latter group, further studies have localized the effect of BMI to the pituitary (50). However, because obesity cannot explain the pituitary resistance to GnRH in all the women in this subgroup, it is also possible that the obese IHH women with pituitary resistance share a unique genetic defect that causes both HH and obesity, as in patients with mutations in leptin (62) and its receptor (63). Of note, in the current study, the GnRH dose was weight-adjusted and was administered iv, making it unlikely that inadequate dosing accounts for the hypogonadotropic responses observed in a number of obese IHH women.
A small group of IHH women responded to GnRH with supraphysiological levels of FSH, implying a degree of ovarian resistance. Dual defects at the hypothalamus and ovary may also explain why none of these women had early signs of pubertal development in contrast to the normal responders and pituitary-resistant groups. The women with ovarian resistance were also normosmic, suggesting that the gene responsible for this IHH subtype is likely to act after GnRH neuronal migration along the olfactory tracts during early embryonic development, although these conclusions are limited by the small number of women with ovarian resistance.
The rate of rise in FSH in response to GnRH replacement in the hypergonadotropic women was identical to that in a 63-year-old IHH woman of postmenopausal age. Although older age may have contributed to the hypergonadotropic response observed in the 41-year-old woman in the ovarian resistance group, age is unlikely to account for the responses of the other 2 women in this group. Furthermore, the control group and the groups of IHH women with normal or hypogonadotropic responses to GnRH also included women in their 30s and early 40s. Because the hypergonadotropic women responded to treatment with higher-dose (or prolonged) GnRH or high-dose exogenous gonadotropins, the secretion of biologically inactive gonadotropins is an unlikely explanation for their results.
Because most genes associated with IHH are expressed in the human ovary (12–16, 18), we were surprised that none of the women with ovarian resistance had a mutation in any of the 14 genes tested. Similarly, genetic defects were absent in most IHH men with testicular resistance (3). Taken together, these studies indicate that there is an additional set of unidentified genes expressed in the hypothalamus that are also relevant for ovarian function. Potential candidates include genes encoding neurosteroidogenic enzymes or intracellular transduction signals.
Physiological replacement of GnRH recreates normal menstrual cycle dynamics in most women with IHH. However, hypogonadotropic responses in the first week of treatment identify a subset of women with pituitary dysfunction, only some of whom have mutations in GNRHR. In a further subset, hypergonadotropic responses to GnRH replacement indicate the presence of dual defects at the hypothalamus and ovary. Identification of many of the genes responsible for IHH has resulted in an explosion of information regarding the central control of reproduction. Investigation of genes with overlapping expression patterns in the hypothalamus and pituitary or ovary in this patient population provides an important strategy for identification of novel genes that are critical for both the central and peripheral control of reproduction.
Acknowledgments
The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Center for Research Resources, or the National Institutes of Health. This study was registered with ClinicalTrials.gov ID# NCT00383656.
This work was supported by the Eunice Kennedy Shriver National Institutes of Child Health and Human Development/National Institutes of Health through R01 HD42708 and cooperative agreement U54 HD028138 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research and M01-RR-01066 National Institutes of Health National Center for Research Resources, General Clinical Research Centers Program. N.D.S. received fellowship support from the National Institutes of Health (Grant 5F32 HD062315) and from the Scholars in Clinical Science program of Harvard Catalyst [The Harvard Clinical and Translational Science Center (Award UL1 RR 025758) and financial contributions from Harvard University and its affiliated academic health care centers].
Disclosure Summary: The authors have nothing to declare.
Footnotes
- BMI
- Body mass index
- CHD7
- chromodomain-helicase-DNA binding protein 7
- E2
- estradiol
- FGFR1
- fibroblast growth factor receptor 1
- GNRHR
- GnRH receptor
- IHH
- isolated hypogonadotropic hypogonadism
- IQR
- interquartile range
- LOF
- loss of function
- P4
- progesterone
- PROK2
- prokineticin 2
- PROKR2
- prokineticin receptor 2
- RSV
- rare sequence variant
- TAC3
- tachykinin 3.
References
- 1. Balasubramanian R, Crowley WF., Jr Isolated GnRH deficiency: a disease model serving as a unique prism into the systems biology of the GnRH neuronal network. Mol Cell Endocrinol. 2011;346(1–2):4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF., Jr Clinical review 15: management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 1990;71(5):1081A–1081G [DOI] [PubMed] [Google Scholar]
- 3. Sykiotis GP, Hoang XH, Avbelj M, et al. Congenital idiopathic hypogonadotropic hypogonadism: evidence of defects in the hypothalamus, pituitary, and testes. J Clin Endocrinol Metab. 2010;95(6):3019–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seminara SB, Beranova M, Oliveira LM, Martin KA, Crowley WF, Jr, Hall JE. Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab. 2000;85(2):556–562 [DOI] [PubMed] [Google Scholar]
- 5. Brinkmeier ML, Davis SW, Carninci P, et al. Discovery of transcriptional regulators and signaling pathways in the developing pituitary gland by bioinformatic and genomic approaches. Genomics. 2009;93(5):449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ericson J, Norlin S, Jessell TM, Edlund T. Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development. 1998;125(6):1005–1015 [DOI] [PubMed] [Google Scholar]
- 7. Ikeda Y, Swain A, Weber TJ, et al. Steroidogenic factor 1 and Dax-1 colocalize in multiple cell lineages: potential links in endocrine development. Mol Endocrinol. 1996;10(10):1261–1272 [DOI] [PubMed] [Google Scholar]
- 8. Lin DC, Bullock CM, Ehlert FJ, Chen JL, Tian H, Zhou QY. Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J Biol Chem. 2002;277(22):19276–19280 [DOI] [PubMed] [Google Scholar]
- 9. McCabe MJ, Gaston-Massuet C, Tziaferi V, et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab. 2011;96(10):E1709–E1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanlaville D, Etchevers HC, Gonzales M, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43(3):211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asakai R, Song SY, Itoh N, Yamakuni T, Tamura K, Okamoto R. Differential gene expression of fibroblast growth factor receptor isoforms in rat ovary. Mol Cell Endocrinol. 1994;104(1):75–80 [DOI] [PubMed] [Google Scholar]
- 12. Cejudo RA, Pinto FM, Dorta I, et al. Analysis of the expression of neurokinin B, kisspeptin, and their cognate receptors NK3R and KISS1R in the human female genital tract. Fertil Steril. 2012;97(5):1213–1219 [DOI] [PubMed] [Google Scholar]
- 13. Choi JH, Gilks CB, Auersperg N, Leung PC. Immunolocalization of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and type I GnRH receptor during follicular development in the human ovary. J Clin Endocrinol Metab. 2006;91(11):4562–4570 [DOI] [PubMed] [Google Scholar]
- 14. Ferrara N, Frantz G, LeCouter J, et al. Differential expression of the angiogenic factor genes vascular endothelial growth factor (VEGF) and endocrine gland-derived VEGF in normal and polycystic human ovaries. Am J Pathol. 2003;162(6):1881–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaytan F, Gaytan M, Castellano JM, et al. KiSS-1 in the mammalian ovary: distribution of kisspeptin in human and marmoset and alterations in KiSS-1 mRNA levels in a rat model of ovulatory dysfunction. Am J Physiol Endocrinol Metab. 2009;296(3):E520–E531 [DOI] [PubMed] [Google Scholar]
- 16. Ghosh P, Saha SK, Bhattacharya S, Bhattacharya S, Mukherjee S, Roy SS. Tachykinin family genes and their receptors are differentially expressed in the hypothyroid ovary and pituitary. Cell Physiol Biochem. 2007;20(5):357–368 [DOI] [PubMed] [Google Scholar]
- 17. Hu Y, Yu H, Shaw G, Pask AJ, Renfree MB. Kallmann syndrome 1 gene is expressed in the marsupial gonad. Biol Reprod. 2011;84(3):595–603 [DOI] [PubMed] [Google Scholar]
- 18. Sato Y, Suzuki T, Hidaka K, et al. Immunolocalization of nuclear transcription factors, DAX-1 and COUP-TF II, in the normal human ovary: correlation with adrenal 4 binding protein/steroidogenic factor-1 immunolocalization during the menstrual cycle. J Clin Endocrinol Metab. 2003;88(7):3415–3420 [DOI] [PubMed] [Google Scholar]
- 19. Valve E, Penttila TL, Paranko J, Harkonen P. FGF-8 is expressed during specific phases of rodent oocyte and spermatogonium development. Biochem Biophys Res Commun. 1997;232(1):173–177 [DOI] [PubMed] [Google Scholar]
- 20. Shaw ND, Seminara SB, Welt CK, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab. 2011;96(3):E566–E576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doty RL, Shaman P, Kimmelman CP, Dann MS. University of Pennsylvania Smell Identification Test: a rapid quantitative olfactory function test for the clinic. Laryngoscope. 1984;94(2 Pt 1):176–178 [DOI] [PubMed] [Google Scholar]
- 22. Hayes FJ, McNicholl DJ, Schoenfeld D, Marsh EE, Hall JE. Free α-subunit is superior to luteinizing hormone as a marker of gonadotropin-releasing hormone despite desensitization at fast pulse frequencies. J Clin Endocrinol Metab. 1999;84(3):1028–1036 [DOI] [PubMed] [Google Scholar]
- 23. Santen RJ, Bardin CW. Episodic luteinizing hormone secretion in man. Pulse analysis, clinical interpretation, physiologic mechanisms. J Clin Invest. 1973;52(10):2617–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng MY, Bullock CM, Li C, et al. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature. 2002;417(6887):405–410 [DOI] [PubMed] [Google Scholar]
- 25. Cole LW, Sidis Y, Zhang C, et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab. 2008;93(9):3551–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Roux N, Young J, Misrahi M, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337(22):1597–1602 [DOI] [PubMed] [Google Scholar]
- 27. Falardeau J, Chung WC, Beenken A, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118(8):2822–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gianetti E, Tusset C, Noel SD, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95(6):2857–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lalani SR, Safiullah AM, Fernbach SD, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78(2):303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miura K, Acierno JS, Jr, Seminara SB. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J Hum Genet. 2004;49(5):265–268 [DOI] [PubMed] [Google Scholar]
- 31. Oliveira LM, Seminara SB, Beranova M, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86(4):1532–1538 [DOI] [PubMed] [Google Scholar]
- 32. Pitteloud N, Zhang C, Pignatelli D, et al. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 2007;104(44):17447–17452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349(17):1614–1627 [DOI] [PubMed] [Google Scholar]
- 34. Beranova M, Oliveira LM, Bedecarrats GY, et al. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2001;86(4):1580–1588 [DOI] [PubMed] [Google Scholar]
- 35. Costa EM, Bedecarrats GY, Mendonca BB, Arnhold IJ, Kaiser UB, Latronico AC. Two novel mutations in the gonadotropin-releasing hormone receptor gene in Brazilian patients with hypogonadotropic hypogonadism and normal olfaction. J Clin Endocrinol Metab. 2001;86(6):2680–2686 [DOI] [PubMed] [Google Scholar]
- 36. Meysing AU, Kanasaki H, Bedecarrats GY, et al. GNRHR mutations in a woman with idiopathic hypogonadotropic hypogonadism highlight the differential sensitivity of luteinizing hormone and follicle-stimulating hormone to gonadotropin-releasing hormone. J Clin Endocrinol Metab. 2004;89(7):3189–3198 [DOI] [PubMed] [Google Scholar]
- 37. Pitteloud N, Quinton R, Pearce S, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 2007;117(2):457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sunyaev S, Ramensky V, Koch I, Lathe W, III, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10(6):591–597 [DOI] [PubMed] [Google Scholar]
- 39. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–576 [DOI] [PubMed] [Google Scholar]
- 40. Thomas PD, Kejariwal A, Campbell MJ, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31(1):334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferrer-Costa C, Gelpi JL, Zamakola L, et al. a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21(14):3176–3178 [DOI] [PubMed] [Google Scholar]
- 43. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor AE, Whitney H, Hall JE, Martin K, Crowley WF., Jr Midcycle levels of sex steroids are sufficient to recreate the follicle-stimulating hormone but not the luteinizing hormone midcycle surge: evidence for the contribution of other ovarian factors to the surge in normal women. J Clin Endocrinol Metab. 1995;80(5):1541–1547 [DOI] [PubMed] [Google Scholar]
- 45. Welt CK, Adams JM, Sluss PM, Hall JE. Inhibin A and inhibin B responses to gonadotropin withdrawal depends on stage of follicle development. J Clin Endocrinol Metab. 1999;84(6):2163–2169 [DOI] [PubMed] [Google Scholar]
- 46. Welt CK, Falorni A, Taylor AE, Martin KA, Hall JE. Selective theca cell dysfunction in autoimmune oophoritis results in multifollicular development, decreased estradiol, and elevated inhibin B levels. J Clin Endocrinol Metab. 2005;90(5):3069–3076 [DOI] [PubMed] [Google Scholar]
- 47. Filicori M, Santoro N, Merriam GR, Crowley WF., Jr Characterization of the physiological pattern of episodic gonadotropin secretion throughout the human menstrual cycle. J Clin Endocrinol Metab. 1986;62(6):1136–1144 [DOI] [PubMed] [Google Scholar]
- 48. Taylor AE, McCourt B, Martin KA, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82(7):2248–2256 [DOI] [PubMed] [Google Scholar]
- 49. Arroyo A, Laughlin GA, Morales AJ, Yen SS. Inappropriate gonadotropin secretion in polycystic ovary syndrome: influence of adiposity. J Clin Endocrinol Metab. 1997;82(11):3728–3733 [DOI] [PubMed] [Google Scholar]
- 50. Pagan YL, Srouji SS, Jimenez Y, Emerson A, Gill S, Hall JE. Inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome: investigation of hypothalamic and pituitary contributions. J Clin Endocrinol Metab. 2006;91(4):1309–1316 [DOI] [PubMed] [Google Scholar]
- 51. Chikazawa K, Araki S, Tamada T. Morphological and endocrinological studies on follicular development during the human menstrual cycle. J Clin Endocrinol Metab. 1986;62(2):305–313 [DOI] [PubMed] [Google Scholar]
- 52. Gross KM, Matsumoto AM, Bremner WJ. Differential control of luteinizing hormone and follicle-stimulating hormone secretion by luteinizing hormone-releasing hormone pulse frequency in man. J Clin Endocrinol Metab. 1987;64(4):675–680 [DOI] [PubMed] [Google Scholar]
- 53. Raivio T, Sidis Y, Plummer L, et al. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2009;94(11):4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoffman AR, Crowley WF., Jr Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982;307(20):1237–1241 [DOI] [PubMed] [Google Scholar]
- 55. Hadziselimovic F, Thommen L, Girard J, Herzog B. The significance of postnatal gonadotropin surge for testicular development in normal and cryptorchid testes. J Urol. 1986;136(1 Pt 2):274–276 [DOI] [PubMed] [Google Scholar]
- 56. Gianetti E, Hall JE, Au MG, et al. When genetic load does not correlate with phenotypic spectrum: lessons from the GnRH receptor (GNRHR). J Clin Endocrinol Metab. 2012;97(9):E1798–E1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raivio T, Avbelj M, McCabe MJ, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012;97(4):E694–E699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reynaud R, Jayakody SA, Monnier C, et al. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab. 2012;97(6):E1068–E1073 [DOI] [PubMed] [Google Scholar]
- 59. Kim HG, Kurth I, Lan F, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83(4):511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Layman WS, Hurd EA, Martin DM. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum Mol Genet. 2011;20(16):3138–3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. St-Amand J, Yoshioka M, Tanaka K, Nishida Y. Transcriptome-wide identification of preferentially expressed genes in the hypothalamus and pituitary gland. Front Endocrinol (Lausanne). 2011;2:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;84(10):3686–3695 [DOI] [PubMed] [Google Scholar]
- 63. Clement K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392(6674):398–401 [DOI] [PubMed] [Google Scholar]