

Abstract
Purpose
To assess therapy-related acute myeloid leukemia/myelodysplastic syndrome (t-AML/MDS) risk in patients treated for Hodgkin lymphoma (HL) on successive generations of Stanford clinical trials.
Patients and Methods
Patients with HL treated at Stanford with at least 5 years of follow-up after completing therapy were identified from our database. Records were reviewed for outcome and development of t-AML/MDS.
Results
Seven hundred fifty-four patients treated from 1974 to 2003 were identified. Therapy varied across studies. Radiotherapy evolved from extended fields (S and C studies) to involved fields (G studies). Primary chemotherapy was mechlorethamine, vincristine, procarbazine, and prednisone (MOPP) or procarbazine, mechlorethamine, and vinblastine (PAVe) in S studies; MOPP, PAVe, vinblastine, bleomycin, and methotrexate (VBM), or doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) in C studies; and VbM (reduced dose of bleomycin compared with VBM) or mechlorethamine, doxorubicin, vinblastine, vincristine, bleomycin, etoposide, and prednisone (Stanford V) in G studies. Cumulative exposure to alkylating agent (AA) was notably lower in the G studies compared with the S and C studies, with a 75% to 83% lower dose of nitrogen mustard in addition to omission of procarbazine and melphalan. Twenty-four (3.2%) of 754 patients developed t-AML/MDS, 15 after primary chemotherapy and nine after salvage chemotherapy for relapsed HL. The incidence of t-AML/MDS was significantly lower in the G studies (0.3%) compared with the S (5.7%) or C (5.2%) studies (P < .001). Additionally, in the G studies, no t-AML/MDS was noted after primary therapy, and the only patient who developed t-AML/MDS did so after second-line therapy.
Conclusion
Our data demonstrate the relationship between the cumulative AA dose and t-AML/MDS. Limiting the dose of AA and decreased need for secondary treatments have significantly reduced the incidence of t-AML/MDS, which was extremely rare in the G studies (Stanford V era).
INTRODUCTION
Secondary malignancies have long been recognized as a complication of treatment for Hodgkin lymphoma (HL).1–3 Therapy-related acute myeloid leukemia (t-AML)/myelodysplastic syndrome (MDS) is an important subset associated with exposure to alkylating agents (AAs) and topoisomerase II inhibitors and with a poor prognosis.4–14 The relative risk of AA-related leukemia peaks in the first 5 years after therapy and is associated with dysplastic changes and chromosomal aberrations involving chromosomes 5 and 7.15,16 In contrast, epipodophyllotoxin-related t-AML occurs 2 to 3 years after exposure, generally lacks a preceding MDS phase, and is characterized by balanced translocations involving 11q23 and 21q22.8,14,16 Other risk factors variably attributed to t-AML/MDS include extended-field radiation therapy (RT), combined-modality therapy, splenectomy, age (> 40 years old), and advanced-stage disease.5,17 The role of involved-field RT (IFRT) as a risk factor has been controversial, and recent large studies suggest no increased risk.15,18
Over the last three decades, improved imaging modalities and recognition of clinical risk factors have replaced the routine use of staging laparotomy and splenectomy, practiced in the 1970s and early 1980s.19–21 Therapy has been modified from intense AA-based chemotherapy to minimal or non–AA-based chemotherapy along with significant modifications of RT fields and doses (ie, high-dose [≥ 44 Gy] extended-field RT to as little as 20 Gy of IFRT).21–24 In this study, we report on the impact of these modifications on the risk of developing t-AML/MDS over three generations of clinical trials for HL at Stanford University Medical Center.
PATIENTS AND METHODS
Patient Selection
This retrospective analysis identified from our HL database patients treated with RT alone, chemotherapy alone, or combined-modality therapy and observed at Stanford over the last three decades.
Methods
The diagnosis of HL was confirmed in the laboratory of Surgical Pathology at Stanford, and the Ann Arbor classification system was used for initial staging.25 All patients had a chest x-ray, CBC, metabolic panel, and erythrocyte sedimentation rate. A bone marrow biopsy was performed in all patients with “B” symptoms or subdiaphragmatic disease and repeated at the end of therapy if involved. Radiographic studies varied over the different eras and included lymphography, computed tomography, gallium scanning, and fluorodeoxyglucose-positron emission tomography– computed tomography. Staging laparotomy with splenectomy, performed in the 1970s and early 1980s, was abandoned as other factors predictive for occult subdiaphragmatic disease were recognized and systemic therapy was incorporated into initial management.26 Follow-up included a physical examination, chest x-ray, CBC, and metabolic panel every 3 months for the first 2 years after treatment, every 6 months for years 3 to 5, and annually thereafter. Era-appropriate imaging was performed annually for the first 3 years or as clinically indicated.
Patients were treated on three generations of treatment protocols, referred to as the S, C, and G studies (Table 1), and patients who developed t-AML/MDS were identified according to the French-American-British classification.27 We also sought to evaluate whether t-AML/MDS developed after primary therapy for HL or after second-line therapy for relapsed disease. The study was conducted in accordance with Stanford's Institutional Review Board and the Declaration of Helsinki.
Table 1.
Patient Demographics and Clinical Characteristics and Chemotherapy and Radiotherapy Characteristics
| Demographic or Characteristic | Study |
||
|---|---|---|---|
| S | C | G | |
| Years of study | 1974-1980 | 1981-1989 | 1989-2003 |
| Patients, No. | 227 | 193 | 334 |
| Median age, years | 27 | 26 | 29 |
| Male | |||
| No. | 129 | 113 | 179 |
| % | 57 | 59 | 54 |
| Median follow-up, years | 22 | 20 | 11 |
| Stage | |||
| I-II | |||
| No. | 96 | 97 | 226 |
| % | 42 | 50 | 68 |
| III-IV | |||
| No. | 131 | 96 | 108 |
| % | 58 | 50 | 32 |
| Splenectomy | |||
| No. | 200 | 122 | 34 |
| % | 88 | 63 | 10 |
| Cumulative doses of chemotherapy agents, mg/m2 | |||
| Mechlorethamine* | 72 | 72 | 12-18 |
| Procarbazine† | 8,400 | 8,400 | — |
| Melphalan‡ | 180 | 180 | — |
| Etoposide | — | — | 240-360 |
| RT dose in S, Gy | |||
| Stage I-II, IF | 44 | ||
| Stage III, STLI/TLI | 44 | ||
| Stage IV, IF or STLI/TLI | 44 | ||
| RT dose in C, Gy | |||
| Stage I-II, IF | 40-44 | ||
| Stage IIX, mantle | 44 | ||
| Stage III-IV, IF or STLI/TLI | 30-44 | ||
| RT dose in G, Gy | |||
| Stage I-II, modified IF | 30-36 | ||
| Stage IIX, III-IV, modified IF, sites > 5 cm, spleen | 36 | ||
NOTE. S studies used MOPP or PAVe; C studies used MOPP, PAVe, ABVD, or VBM; and G studies used VbM or Stanford V.
Abbreviations: ABVD, doxorubicin, bleomycin, vinblastine, dacarbazine; IF, involved field; MOPP, mechlorethamine, vincristine, procarbazine, prednisone; PAVe, procarbazine, melphalan, vinblastine; RT, radiation therapy; Stanford V, mechlorethamine, doxorubicin, vinblastine, vincristine, bleomycin, etoposide, prednisone; STLI, subtotal lymphoid irradiation; TLI, total lymphoid irradiation; VBM, vinblastine, bleomycin, methotrexate; VbM, VBM but with reduced dose of bleomycin; X, bulky disease.
Mechlorethamine used in MOPP and Stanford V regimens only.
Procarbazine used in MOPP and PAVe only.
Melphalan used in PAVe only.
Specifics of Therapy
The S studies were randomized clinical trials conducted between 1974 and 1980. Patients with stage I or II disease were treated with either RT (44 Gy) as a single modality (subtotal lymphoid irradiation [STLI] or total lymphoid irradiation [TLI]) or combined-modality therapy with IFRT (44 Gy) followed by MOPP mechlorethamine, vincristine, procarbazine, and prednisone (MOPP; Appendix Table A1, online only).28 Patients with stage IIE to IIIB disease received chemotherapy with six cycles of either MOPP or procarbazine, mechlorethamine, and vinblastine (PAVe) combined in an alternating fashion with RT (STLI 44 Gy for stage IIE disease and TLI 44 Gy for stage III disease). Patients with stage IV disease were treated either with MOPP or PAVe plus RT (IFRT or TLI; 44 Gy) delivered in a split-course fashion or with MOPP alone.
The C studies were randomized trials conducted between 1981 and 1989. Patients with stage I or II disease and favorable presentations of stage IIIA disease were treated either with RT alone (40 to 44 Gy STLI/TLI) or IFRT (40 to 44 Gy) followed by six cycles of vinblastine, bleomycin, and methotrexate (VBM) chemotherapy.29 Patients with bulky mediastinal disease were treated with 6 months of PAVe or 6 months of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) combined with 44 Gy of mantle-field RT (stage I or II) or 44 Gy of STLI/TLI (stage III) in a split-course or alternating fashion. Patients with stage IIIB to IV disease were treated with either combined-modality therapy (PAVe + TLI 30 to 44 Gy) or chemotherapy alone (PAVe/ABVD or MOPP/ABVD).
Patients on the G studies were treated between 1989 and 2003. Staging laparotomy was abandoned, and notably, neither MOPP nor PAVe chemotherapy was used. In the G1 trial, patients with nonbulky stage I to IIA disease were randomly assigned between treatment with RT alone (STLI 36 to 44 Gy) or combined-modality therapy with 2 months of VBM followed by mantle-field RT (36 to 40 Gy) and 4 months of VbM (reduced dose of bleomycin compared with VBM). After 1992, all patients were treated with minimal alkylator therapy on the Stanford V regimen (mechlorethamine, doxorubicin, vinblastine, vincristine, bleomycin, etoposide, and prednisone) for 8 or 12 weeks depending on stage. This was followed by IFRT (30 Gy) in nonbulky stage I to IIA disease or 36 Gy to sites greater than 5 cm and to involved spleen in stage IIX, III, and IV disease.29,30 RT field modifications were also notable for the omission of the high neck and axillae from the RT field unless the latter sites were involved with disease.29 This represented a considerable reduction in RT dose and field size compared with the S and C studies.
Statistical Analysis
All statistical analyses were conducted using SAS Enterprise Guide 4.3 and SAS 9.3 (SAS Institute, Cary, NC). Comparisons of relative incidences of t-AML/MDS across the three treatment eras were made. Progression-free survival (PFS) was defined as time to treatment failure, measured as time from entry onto the study to disease progression, treatment failure, or death. Overall survival (OS) was defined as time from study entry until death from any cause.31 For the mortality analysis, living patients were censored at the time of last follow-up. For the t-AML/MDS analysis, deceased patients who did not have leukemia were censored at date of death or last contact. Kaplan-Meier methods with log-rank tests were used to evaluate PFS, OS, and percentage of patients free of t-AML/MDS.32 Comparisons between treatment groups for the log-rank test were adjusted for multiple comparisons using Tukey-Kramer methods.33 Cox regression methods were used to assess the impact of treatment modality while controlling for age and stage at disease presentation.34 Freeman-Halton tests (an extension of Fisher's exact test) and Kruskal-Wallis tests were used to compare demographics, disease characteristics, and t-AML/MDS incidence across studies.35,36
RESULTS
Seven hundred fifty-four patients treated between 1974 and 2003 who met predefined study criteria were identified. Patient characteristics and therapy details are listed in Table 1. As expected, statistically significant differences were observed in median follow-up (driven by the shorter time in the more recent G studies) and splenectomy (staging laparotomy was rarely performed in the G era). The distributions by stage and age were also statistically different. More patients in the G studies had early-stage disease and were slightly older compared with the S and C studies. Fifty-five percent of patients in the G studies had stage II disease, of which 35% were considered bulky and treated as advanced disease. Follow-up time of greater than 7 years was available for 87%, 80%, and 87% of patients in the S, C, and G studies, respectively.
Comparison of Treatment Regimens and Outcomes
In the S studies, 12 patients (5%) were treated with chemotherapy alone, 35 (15%) with RT alone, and the rest with combined-modality therapy. Chemotherapy consisted of MOPP in 130 patients (57%) and PAVe in 62 patients (27%). RT consisted of STLI/TLI in 148 patients (65%) and IFRT in 67 patients (30%). The median follow-up time is 22 years. The 10-year PFS and OS were 67% and 77%, respectively, and the 20-year PFS and OS were 57% and 64%, respectively (Table 2, Figs 1 and 2). In the C studies, 38 patients (20%) were treated with RT alone, and the rest were treated with combined-modality therapy. Ninety-eight patients (51%) received AA-based chemotherapy (ie, MOPP or PAVe) without RT (41%), with IFRT (24%), or with STLI/TLI (35%). Fifty-seven patients (37%) received non–AA-based chemotherapy (ABVD or VBM) with IFRT. The median follow-up time is 20 years. The 10-year PFS and OS were 71% and 82%, respectively, and the 20-year PFS and OS were 62% and 70%, respectively (Table 2, Figs 1 and 2). A majority of patients in the G studies were treated with combined-modality therapy with either VBM/VbM (n = 35, 10%) or the Stanford V regimen (n = 256, 77%). The median follow-up time is 11 years. The 10-year PFS and OS were 88% and 94%, respectively (Table 2, Figs 1 and 2).
Table 2.
Outcome and Incidence of t-AML/MDS in Each Era of Therapy After Primary or Second-Line Treatment
| Study | No. of Patients | 10 Years |
20 Years | Patients With t-AML/MDS |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| After Primary Therapy |
After Second-Line Therapy |
Total No. | ||||||||
| PFS (%) | OS (%) | PFS (%) | OS (%) | No./Total No. | % | No./Total No. | % | |||
| S | 227 | 67 | 77 | 57 | 64 | 9/170 | 5 | 4/57 | 7 | 13 |
| C | 193 | 71 | 82 | 62 | 70 | 6/147 | 4 | 4/46 | 9 | 10 |
| G | 334 | 88 | 94 | NA | NA | 0/299 | 0 | 1/35 | 3 | 1 |
Abbreviations: NA, not applicable; OS, overall survival; PFS, progression-free survival; t-AML/MDS, therapy-related acute myeloid leukemia/myelodysplastic syndrome.
Fig 1.

Kaplan-Meier progression-free survival for the G, C, and S studies.
Fig 2.

Kaplan-Meier overall survival for the G, C, and S studies.
Overall, 138 patients (18.3%) experienced relapse. More relapses were noted in patients with stage III or IV disease in the S (n = 39, 61%) and C studies (n = 30, 65%) versus the G studies (n = 16, 45%). This improvement is reflected in PFS and OS in the G studies compared with the C and S studies (P < .001; Figs 1 and 2).
Because there were baseline differences in stage, age, and splenectomy across treatment eras, multivariate Cox models were built using the latter factors and treatment era (S, C, or G). In these models, splenectomy was not statistically significant; however, treatment regimen remained statistically significant for OS, PFS, and the incidence of t-AML/MDS after controlling for both age and stage (Appendix Tables A2 and A3 and Appendix Figs A1A and A1B, online only).
Comparison of Chemotherapy Doses Across S, C, and G Studies
The cumulative doses of AA and the topoisomerase II inhibitor etoposide for each regimen are listed in Table 1. In the S and C studies, patients treated with six cycles of MOPP received cumulative doses of mechlorethamine 72 mg/m2 and procarbazine 8,400 mg/m2, whereas patients treated with six cycles of PAVe received cumulative doses of 180 mg/m2 and 8,400 mg/m2 of melphalan and procarbazine, respectively. In contrast, in the G studies, patients treated with Stanford V received cumulative doses of 12 or 18 mg/m2 of mechlorethamine and 240 or 360 mg/m2 of etoposide on the 8- or 12-week regimen, respectively. Therefore, in the G studies, there was a substantial (75% to 83%) reduction in total mechlorethamine dose compared with that delivered with six cycles of MOPP, as well as the omission of procarbazine and melphalan.
Comparison of RT Doses and Fields Across S, C, and G Studies
RT doses and fields across different stages of disease for each of the S, C, and G studies are listed in Table 1. The most notable difference between the studies is the evolution of RT from doses of 44 Gy delivered to STLI or TLI fields in the S and C studies to significantly lower doses of 30 to 36 Gy delivered to a modified involved field for stage I and II disease or to sites of disease more than 5 cm or macroscopic splenic disease in stage IIX, III, or IV in the G studies.
Comparison of t-AML/MDS Across S, C,and G Studies
Overall, 24 (3.2%) of 754 patients developed t-AML/MDS, including 13 patients (5.7%) in S studies, 10 patients (5.2%) in C studies, and one patient (0.3%) in G studies (Table 2). The median age at diagnosis of t-AML/MDS was 41 years (range, 21 to 70 years). Although there was no difference in t-AML/MDS risk between the S and C studies (P = .86), a significant decrease in risk was noted in the G studies (G v S, P = .002; G v C, P = .007; Fig 3A). After primary therapy, 15 patients (all in the S or C studies) developed t-AML/MDS at a median of 4.4 years (range, 1.9 to 18 years; Fig 3B). Of the 138 patients who experienced relapse and were treated with additional AAs as part of salvage therapy, nine patients developed t-AML/MDS (S, n = 4; C, n = 4; G, n = 1) at a median of 4.7 years (range, 2.2 to 10.8 years; Tables 2 and 3). Late development of t-AML/MDS more than 15 years after primary therapy was observed in two patients (one each in the S and C studies). Table 3 lists the characteristics of the 24 patients who developed t-AML/MDS. Notably, the majority of patients (20 of 24 patients, 83%) had received chemotherapy with either MOPP or PAVe, whereas only one patient developed t-AML/MDS after ABVD alone. None of the patients treated with VbM or Stanford V developed t-AML/MDS after primary therapy. Of the 116 patients treated with RT alone, two patients treated with STLI/TLI in the C studies developed t-AML/MDS (in one patient after additional AA therapy for relapse).
Fig 3.
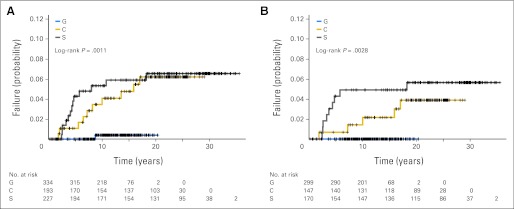
(A) Incidence of therapy-related acute myeloid leukemia/myelodysplastic syndrome (t-AML/MDS) in the G, C, and S studies. (B) Incidence of t-AML/MDS after primary therapy in the G, C, and S studies.
Table 3.
Characteristics of Patients With t-AML/MDS
| Patient No. | Stage at Diagnosis | Primary CT | Primary RT | RT Dose (Gy) | Secondary CT for Relapse | Time to t-AML/MDS After Primary Therapy (years) | Time to t-AML/MDS After Secondary Therapy (years) | Time From t-AML/MDS to Death (years) | FAB Subtype/Cytogenetics |
|---|---|---|---|---|---|---|---|---|---|
| 1 | IVA | MOPP | None | BCAVe | 4.1 | 3.1 | 1.3 | AML NOS | |
| 2 | IIIAS | MOPP | TLI | 44 | None | 2.6 | NA | 0.7 | M4 |
| 3 | IVB | MOPP | None | None | 3.3 | NA | 0.6 | AML NOS | |
| 4 | IIIB | PAVe | TLI | 44 | ABVD/MOPP | 10.7 | 1.8 | 0.6 | M4 |
| 5 | IVB | MOPP | TLI | 44 | ABVD, chlorambucil, CEOP, RT | 8 | 6.6 | 0.6 | AML NOS |
| 6 | IIA | MOPP | IF | 44 | None | 3.7 | NA | 0.6 | AML NOS |
| 7 | IIIAS | MOPP | TLI | 44 | None | 4.4 | NA | 0.7 | M4 |
| 8 | IIA | MOPP | IF | 44 | BCAVe, RT | 4.6 | 1.5 | 1.7 | M4 |
| 9 | IIIBS | MOPP | TLI | 44 | None | 2.4 | NA | 1.6 | AML NOS |
| 10 | IVA | MOPP | IF | 44 | None | 2.6 | NA | 0.7 | AML NOS |
| 11 | IIIBS | MOPP | TLI | 44 | None | 4.7 | NA | 0.7 | AML NOS |
| 12 | IIIAS | MOPP | TLI | 44 | None | 5.8 | NA | 1 | AML NOS |
| 13 | IIIBS | MOPP | TLI | 44 | None | 18.2 | NA | 0 | AML NOS* |
| 14 | IIIAES | ABVD/PAVe | None | None | 2.1 | NA | 0.3 | AML NOS | |
| 15 | IIIAS | PAVe | TLI | 44 | ABVD, MOPP, cyclophosphamide, chlorambucil, etoposide | 5.6 | 3.6 | 0.1 | MDS |
| 16 | IIIA | ABVD | IF | 44 | None | 10 | NA | 1.2 | M1/46XY, inv(9)(p11q13) |
| 17 | IVB | ABVD/PAVe | None | Cyclophosphamide, chlorambucil, MOPP, etoposide, CHAD, BMT | 6.5 | 4.7 | 1.1 | M4*/45XY, t(13;18)(q14;q11.2), 7− | |
| 18 | IVB | PAVe | TLI | 44 | None | 1.8 | NA | 0.9 | MDS |
| 19 | IVB | ABVD/MOPP | EF | 15 | None | 15.9 | NA | 1.4 | M0/7q−, t(15;17) |
| 20 | IIIA | PAVe | IF | 44 | None | 7.2 | NA | Alive | M5/45X,Y−, t(8;21)(q22;q22) |
| 21 | IVB | ABVD/MOPP | EF | 30 | PAVe | 13.5 | 13.2 | 0.3 | AML NOS* |
| 22 | IIA | NA | EF | 44 | NA | 17 | NA | 0.2 | M4/MLD* |
| 23 | IB | NA | EF | 44 | MOPP, ABVD, Stanford V, CHAD, BMT | 8.2 | 6.3 | 1.7 | MDS |
| 24 | IIA | Stanford V, 12 weeks | Modified IF | 36 | ABVD, MOPP, BMT | 8.7 | 4.9 | Alive | M4/9q−, 11q23 (MLL) |
Abbreviations: A, absence of “B” symptoms; ABVD, doxorubicin, bleomycin, vinblastine, dacarbazine; B, presence of B symptoms; BCAVe, bleomycin, lomustine, doxorubicin, vinblastine; BMT, bone marrow transplantation; CEOP, cyclophosphamide, etoposide, vincristine, prednisone; CHAD, cisplatin, high-dose cytarabine, dexamethasone; CT, chemotherapy; EF, extended field; FAB, French-American-British; IF, involved field; MLD, multilineage dysplasia; MOPP, mechlorethamine, vincristine, procarbazine, prednisone; NA, not applicable; NOS, not otherwise specified in chart; PAVe, procarbazine, melphalan, vinblastine; RT, radiation therapy; S, splenic involvement; Stanford V, mechlorethamine, doxorubicin, vinblastine, vincristine, bleomycin, etoposide, prednisone; STLI, subtotal lymphoid irradiation; t-AML/MDS, therapy-related acute myeloid leukemia/myelodysplastic syndrome; TLI, total lymphoid irradiation.
AML preceded by MDS.
Documentation of cytopenias or myelodysplasia was noted in four patients, perhaps suggestive of a myelodysplastic phase before the diagnosis. The outcome of patients with t-AML/MDS was extremely poor, and 22 patients died as a result of disease. The median time to death once t-AML/MDS was diagnosed was 0.7 years (range, 0 to 1.7 years). Two patients are still alive, both treated with cytoreductive therapy, followed by allogeneic bone marrow transplantation with a syngeneic donor in one patient and a matched unrelated donor in the other.
DISCUSSION
Our retrospective analysis reports 3.2% incidence of t-AML/MDS with a significantly lower risk in the most recent G studies (0.3%) compared with the older S and C studies (5.7%, P = .002; and 5.2%, P = .007, respectively). The median time to development of t-AML/MDS was 4.6 years, consistent with other reports of t-AML/MDS as a result of AA-based chemotherapy.15 It is questionable whether the two cases of t-AML/MDS that developed 15 years after treatment were truly therapy-related or de novo events. Unfortunately, because of the retrospective nature of our study spanning more than three decades, essential cytogenetic studies that may have clarified the latter were not available in most patients.37,38
The risk of developing t-AML/MDS after AA-based chemotherapy is well established, and relative risks have been reported with respect to individual agents and regimens.1,8,13,39 In a multivariate model that examined AA-heavy and AA-poor regimens, the relative risk of t-AML/MDS with MOPP was 5.9, whereas that of ABVD was significantly lower at 1.5.1 A major difference between the chemotherapy regimens used in the S, C, and G studies relates to the cumulative dose of AAs used (Table 1). In the G studies (Stanford V regimen), melphalan and procarbazine were eliminated along, with an 83% reduction in mechlorethamine compared with the C and S studies. Not surprisingly, more than 80% of the patients who developed t-AML/MDS had been treated on one of the S or C studies. The incidence of t-AML/MDS in the G series is also significantly lower than some of the more recent regimens like escalated bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone, which has a 2.5% incidence of t-AML/MDS after primary treatment likely a result of the high doses of cyclophosphamide (9.6 gm/m2) and etoposide (4,800 mg/m2) used.40,41
The risk of developing t-AML/MDS is related not only to the primary therapy, but also to therapy at relapse. Several studies indicate that the number of cycles of chemotherapy to achieve remission correlates with the risk of secondary leukemia.4,12 In our data set, significantly fewer patients required salvage chemotherapy in the G studies (35 of 334 patients, 10%) compared with the earlier S (57 of 227 patients, 25%) and C studies (46 of 193 patients, 24%) which led to a decrease in the incidence of t-AML/MDS in the G studies. Our observations are similar to reports by the German Hodgkin Study Group, in which the incidence of t-AML/MDS across nine trials between 1978 and 1998 decreased to 1% (46 of 5,411 patients), compared with reports of 2.2% to 5.2% when AA-heavy regimens were used.9,12,20,42
Although we observed no cases of t-AML/MDS after primary therapy with the Stanford V regimen, two cases have been reported in the literature. In an Eastern Cooperative Oncology Group pilot study (E1492), one of 47 patients developed t-AML/MDS [M5 subtype, t(9;11),+8] 2 years after treatment.43 Another case was reported from Italy of a patient who developed t-AML/MDS (M3 subtype) 4 years after completing therapy with the Stanford V regimen.44 Data suggest that exposure to topoisomerase II inhibitors (ie, etoposide) and doxorubicin may result in tightly clustered break points in an 8–base pair region in intron 6 of PML-RARA (retinoic acid receptor α) that is associated with the characteristic translocation t(15;17) seen in acute promyelocytic leukemia (M3 type of AML).45 In our series, the patient who developed t-AML/MDS after Stanford V and salvage therapy had karyotypic abnormalities, including 9q− and 11q23 (MLL gene rearrangement), which have also been previously described in patients treated with topoisomerase II inhibitors.
With respect to radiation exposure, two patients treated with STLI/TLI as primary therapy developed t-AML/MDS. However, one did so after 17 years (making a de novo event more likely), and the other developed t-AML/MDS after salvage chemotherapy that included AAs. The additional relative risk of RT to chemotherapy in the development of t-AML/MDS has been discordant in the literature. A meta-analysis including 1,183 patients across 12 trials reported a statistically significant increase in the incidence of leukemia-related deaths in patients receiving combined-modality therapy.46 In another meta-analysis, a nonstatistically significant trend toward increase in t-AML/MDS was seen when comparing chemotherapy with combined-modality therapy across 16 studies (hazard ratio, 1.82; 95% CI, 0.95 to 3.46; P = .07), with no differences between IFRT and STLI/TLI.47,48 In our studies, 12 (7%) of 172 patients developed t-AML/MDS after STLI/TLI (44 Gy) + AAs, five (7.7%) of 65 patients after IFRT (44 Gy) + AAs, and three (5.8%) of 52 patients after AA-based therapy alone, suggesting that the RT per se in combined-modality therapy did not add to the risk conferred by chemotherapy alone. Because only one patient treated on Stanford V developed t-AML/MDS, the additional impact of a lower dose of IFRT on a significantly reduced alkylator regimen cannot be determined.
In conclusion, successive generations of HL treatments at Stanford, specifically the Stanford V regimen, demonstrate a significantly lower risk of t-AML/MDS without compromising efficacy, which was one of the important goals in the development of the latter regimen. The prognosis for t-AML/MDS is dismal, and it is imperative that therapies minimize these risks. Current ongoing adaptive strategies based on interim positron emission tomography imaging to evaluate early response to treatment may allow for selection of patients so that AA exposure can be limited to those with high-risk disease.49 As outcomes continue to improve with modern approaches in management of HL, anticipated cure rates must be balanced with the unintended sequelae of therapy.
Appendix
Table A1.
Details of Study Regimens
| S Studies |
C Studies |
G Studies |
|||
|---|---|---|---|---|---|
| Protocol | Description | Protocol | Description | Protocol | Description |
| s1a | STLI (stg I/IIA) | c1a | STLI (ps I-IIA) | g1a | ST+Spleen |
| s1b | IF+MOP (stg I/IIA) | c1b | IF + VBM×6 (ps I-IIA) | g1a-K | Kaiser ST+Spleen |
| s2a | STLI+MOP (stg IEA/IIEA) | c2a | STLI (ps I-IIB) | g1b | VBM×2/mantle/VBM×4 |
| s2b | STLI+PAVe (stg IEA/IIEA) | c2b | IF + VBM×6 (ps I-IIB) | g1b-K | Kaiser VBM×2/mantle/VBMx4 |
| s3a | TLI+MOP (stg IEB/IIEB) | c3a | TLI (ps IIIA) | g2 | Stanford V×3, consolidated RT (36 Gy) |
| s3b | TLI+PAVe (stg IEB/IIEB) | c3b | IF + VBM×6 (ps IIIA) | g2b | Stanford V, RT |
| s4a | TLI+MOP (stg IIIA/IIISA) | c4a | PAVe×3/mod IF/PAVe×3 | g3 | Stanford V×3, consolidated RT (36 Gy) |
| s4b | TLI+PAVe (stg IIIA/IIISA) | c4b | ABVD×3/mod IF/ABVD×3 | g4 | Stanford V×8 weeks, RT |
| s5a | MOPP-2+TLI+MOPP-4 (stg IIIB | c5a | PAVe×3/mod IF/PAVe×3 | g4-K | Stanford V×8, RT |
| s5b | PAVe-2+TLI+PAVe-4 (stg IIIB | c5b | ABVD×3/mod IF/ABVD×3 | ||
| s6a | MOPP-2+TLI+MOPP-4 (stg IVL) | c6a | PAVe/mod IF ping-pong | ||
| s6b | PAVe-2+TLI+PAVe-4 (stg IVL) | c6b | ABVD/mod IF ping-pong | ||
| s7a | TLI+MOPP-6 (stg IVH) | c7a | PAVe/TLI ping-pong | ||
| s7b | MOPP-6+IF (stg IVH) | c7b | PAVe/ABVD alt × 12 | ||
| s8a | Split-course (stg IVM) | c8a | PAVe/TLI ping-pong | ||
| s8b | MOPP alone | c8b | PAVe/ABVD alt × 12 | ||
| s8b,b | MOPP, bleomycin | c9a | PAVe/TLI ping-pong | ||
| c9b | PAVe/ABVD alt × 12 | ||||
| c10a | PAVe/TLI ping-pong (stg IV) | ||||
| c10b | PAVe/ABVD alt × 12 (stg IV) | ||||
| c12a | PAVe/TLI ping-pong | ||||
| c12b | MOPP/ABVD | ||||
| c13a | PAVe/TLI ping-pong | ||||
| c13b | MOPP/ABVD | ||||
| c14a | PAVe/TLI ping-pong | ||||
| c14b | MOPP/ABVD | ||||
| c15a | PAVe/TLI | ||||
| c15b | MOPP/ABVD | ||||
Abbreviations: ABVD, doxorubicin, bleomycin, vinblastine, dacarbazine; alt, alternate; IF, involved field; mod, modified; MOP, mechlorethamine, vincristine, procarbazine; MOPP, mechlorethamine, vincristine, procarbazine, prednisone; PAVe, procarbazine, melphalan, vinblastine; RT, radiation therapy; ps, pathologic stage; Stanford V, mechlorethamine, doxorubicin, vinblastine, vincristine, bleomycin, etoposide, prednisone; stg, stage; STLI, subtotal lymphoid irradiation; TLI, total lymphoid irradiation; VBM, vinblastine, bleomycin, methotrexate.
Table A2.
Multivariate Analyses of t-AML/MDS Based on Age, Treatment Era, Stage, and Splenectomy
| Treatment Parameter | df | P(χ2) | Hazard Ratio |
|---|---|---|---|
| Age (> v < 28 years) | 1 | .0025 | 1.052 |
| Study (S, C, G) | 2 | .013 | |
| Stage (early v late) | 1 | .0427 | 3.589 |
| Splenectomy | 1 | .0647 | 2.392 |
| Direct comparison for treatment parameter | |||
| Study (S v G) | 1 | .0032 | 25.12 |
| Study (C v G) | 1 | .0071 | 18.582 |
Abbreviation: t-AML/MDS, therapy-related acute myeloid leukemia/myelodysplastic syndrome.
Table A3.
Multivariate Analyses of t-AML/MDS Based on Age, Treatment Era, and Stage
| Treatment Parameter | df | P(χ2) | Hazard Ratio |
|---|---|---|---|
| Age (> v < 28 years) | 1 | .002 | 1.049 |
| Study (S, C, G) | 2 | .0404 | |
| Stage (early v late) | 1 | .0119 | 3.578 |
| Direct comparison for treatment parameter | |||
| Study (S v G) | 1 | .0131 | 13.342 |
| Study (C v G) | 1 | .0131 | 13.633 |
Abbreviation: t-AML/MDS, therapy-related acute myeloid leukemia/myelodysplastic syndrome.
Fig A1.
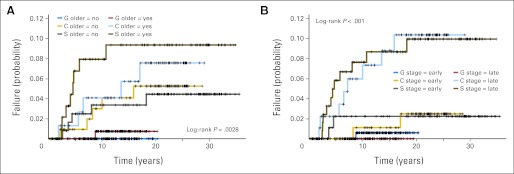
Therapy-related acute myeloid leukemia/myelodysplastic syndrome risk by (A) treatment and age and (B) treatment and Ann Arbor stage early, stage I/II; late, stage III/IV.
Footnotes
Supported by the National Cancer Institute, National Institutes of Health, Grants No. CA56060, CA05838, CA34233, CA09287, and CA21555.
Presented in part at the 42nd Annual Meeting of the American Society of Clinical Oncology, June 2-6, 2006, Atlanta, GA.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Sandra J. Horning, Genentech (C) Consultant or Advisory Role: None Stock Ownership: Sandra J. Horning, Roche Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Provision of study materials or patients: Sandra J. Horning, Saul A. Rosenberg, Richard T. Hoppe, Ranjana H. Advani
Collection and assembly of data: Michael Zach Koontz, Raymond Balise, Ranjana H. Advani
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Boivin JF, Hutchison GB, Zauber AG, et al. Incidence of second cancers in patients treated for Hodgkin's disease. J Natl Cancer Inst. 1995;87:732–741. doi: 10.1093/jnci/87.10.732. [DOI] [PubMed] [Google Scholar]
- 2.van Leeuwen FE, Klokman WJ, Hagenbeek A, et al. Second cancer risk following Hodgkin's disease: A 20-year follow-up study. J Clin Oncol. 1994;12:312–325. doi: 10.1200/JCO.1994.12.2.312. [DOI] [PubMed] [Google Scholar]
- 3.Hoppe RT. Hodgkin's disease: Complications of therapy and excess mortality. Ann Oncol. 1997;8(suppl 1):115–118. [PubMed] [Google Scholar]
- 4.Devereux S, Selassie TG, Vaughan Hudson G, et al. Leukaemia complicating treatment for Hodgkin's disease: The experience of the British National Lymphoma Investigation. BMJ. 1990;301:1077–1080. doi: 10.1136/bmj.301.6760.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin's disease. N Engl J Med. 1990;322:7–13. doi: 10.1056/NEJM199001043220102. [DOI] [PubMed] [Google Scholar]
- 6.Zinzani PL, Fiacchini M, Mazza P, et al. Fifteen-year experience in Hodgkin's disease: Role of combined modality treatment and splenectomy in the incidence of secondary acute leukemia. Haematologica. 1991;76:305–310. [PubMed] [Google Scholar]
- 7.Thirman MJ, Larson RA. Therapy-related myeloid leukemia. Hematol Oncol Clin North Am. 1996;10:293–320. doi: 10.1016/s0889-8588(05)70340-3. [DOI] [PubMed] [Google Scholar]
- 8.Brusamolino E, Anselmo AP, Klersy C, et al. The risk of acute leukemia in patients treated for Hodgkin's disease is significantly higher after combined modality programs than after chemotherapy alone and is correlated with the extent of radiotherapy and type and duration of chemotherapy: A case-control study. Haematologica. 1998;83:812–823. [PubMed] [Google Scholar]
- 9.Josting A, Wiedenmann S, Franklin J, et al. Secondary myeloid leukemia and myelodysplastic syndromes in patients treated for Hodgkin's disease: A report from the German Hodgkin's Lymphoma Study Group. J Clin Oncol. 2003;21:3440–3446. doi: 10.1200/JCO.2003.07.160. [DOI] [PubMed] [Google Scholar]
- 10.Valagussa P, Santoro A, Fossati-Bellani F, et al. Second acute leukemia and other malignancies following treatment for Hodgkin's disease. J Clin Oncol. 1986;4:830–837. doi: 10.1200/JCO.1986.4.6.830. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez MA, Fuller LM, Zimmerman SO, et al. Hodgkin's disease: Study of treatment intensities and incidences of second malignancies. Ann Oncol. 1993;4:125–131. doi: 10.1093/oxfordjournals.annonc.a058414. [DOI] [PubMed] [Google Scholar]
- 12.van Leeuwen FE, Chorus AM, van den Belt-Dusebout AW, et al. Leukemia risk following Hodgkin's disease: Relation to cumulative dose of alkylating agents, treatment with teniposide combinations, number of episodes of chemotherapy, and bone marrow damage. J Clin Oncol. 1994;12:1063–1073. doi: 10.1200/JCO.1994.12.5.1063. [DOI] [PubMed] [Google Scholar]
- 13.Leone G, Mele L, Pulsoni A, et al. The incidence of secondary leukemias. Haematologica. 1999;84:937–945. [PubMed] [Google Scholar]
- 14.Andersen MK, Christiansen DH, Jensen BA, et al. Therapy-related acute lymphoblastic leukaemia with MLL rearrangements following DNA topoisomerase II inhibitors, an increasing problem: Report on two new cases and review of the literature since 1992. Br J Haematol. 2001;114:539–543. doi: 10.1046/j.1365-2141.2001.03000.x. [DOI] [PubMed] [Google Scholar]
- 15.Swerdlow AJ, Higgins CD, Smith P, et al. Second cancer risk after chemotherapy for Hodgkin's lymphoma: A collaborative British cohort study. J Clin Oncol. 2011;29:4096–4104. doi: 10.1200/JCO.2011.34.8268. [DOI] [PubMed] [Google Scholar]
- 16.Pedersen-Bjergaard J, Andersen MK. Secondary or therapy-related MDS and AML and their chromosome aberrations: Important to study but difficult to establish causality. Haematologica. 1998;83:481–482. [PubMed] [Google Scholar]
- 17.Henry-Amar M, Dietrich PY. Acute leukemia after the treatment of Hodgkin's disease. Hematol Oncol Clin North Am. 1993;7:369–387. [PubMed] [Google Scholar]
- 18.Mauch PM, Canellos GP, Rosenthal DS, et al. Reduction of fatal complications from combined modality therapy in Hodgkin's disease. J Clin Oncol. 1985;3:501–505. doi: 10.1200/JCO.1985.3.4.501. [DOI] [PubMed] [Google Scholar]
- 19.Bonadonna G, Bonfante V, Viviani S, et al. ABVD plus subtotal nodal versus involved-field radiotherapy in early-stage Hodgkin's disease: Long-term results. J Clin Oncol. 2004;22:2835–2841. doi: 10.1200/JCO.2004.12.170. [DOI] [PubMed] [Google Scholar]
- 20.Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin's disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med. 1992;327:1478–1484. doi: 10.1056/NEJM199211193272102. [DOI] [PubMed] [Google Scholar]
- 21.Horning SJ, Hoppe RT, Mason J, et al. Stanford-Kaiser Permanente G1 study for clinical stage I to IIA Hodgkin's disease: Subtotal lymphoid irradiation versus vinblastine, methotrexate, and bleomycin chemotherapy and regional irradiation. J Clin Oncol. 1997;15:1736–1744. doi: 10.1200/JCO.1997.15.5.1736. [DOI] [PubMed] [Google Scholar]
- 22.Engert A, Plütschow A, Eich HT, et al. Reduced treatment intensity in patients with early-stage Hodgkin's lymphoma. N Engl J Med. 2010;363:640–652. doi: 10.1056/NEJMoa1000067. [DOI] [PubMed] [Google Scholar]
- 23.Hoppe RT, Horning SJ, Hancock SL, et al. Current Stanford clinical trials for Hodgkin's disease. Recent Results Cancer Res. 1989;117:182–190. doi: 10.1007/978-3-642-83781-4_19. [DOI] [PubMed] [Google Scholar]
- 24.Horning SJ. The Stanford Hodgkin's disease (HD) studies: An update. Leukemia. 1991;5(suppl 1):53–55. [PubMed] [Google Scholar]
- 25.Carbone PP, Kaplan HS, Musshoff K. Report of the Committee on Hodgkin's Disease Staging Classification. Cancer Res. 1971;31:1860–1861. [PubMed] [Google Scholar]
- 26.Carde P, Hagenbeek A, Hayat M, et al. Clinical staging versus laparotomy and combined modality with MOPP versus ABVD in early-stage Hodgkin's disease: The H6 twin randomized trials from the European Organization for Research and Treatment of Cancer Lymphoma Cooperative Group. J Clin Oncol. 1993;11:2258–2272. doi: 10.1200/JCO.1993.11.11.2258. [DOI] [PubMed] [Google Scholar]
- 27.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–199. [PubMed] [Google Scholar]
- 28.Rosenberg SA, Kaplan HS. The evolution and summary results of the Stanford randomized clinical trials of the management of Hodgkin's disease: 1962-1984. Int J Radiat Oncol Biol Phys. 1985;11:5–22. doi: 10.1016/0360-3016(85)90357-8. [DOI] [PubMed] [Google Scholar]
- 29.Horning SJ, Hoppe RT, Breslin S, et al. Stanford V and radiotherapy for locally extensive and advanced Hodgkin's disease: Mature results of a prospective clinical trial. J Clin Oncol. 2002;20:630–637. doi: 10.1200/JCO.2002.20.3.630. [DOI] [PubMed] [Google Scholar]
- 30.Horning SJ, Hoppe RT, Hancock SL, et al. Vinblastine, bleomycin, and methotrexate: An effective adjuvant in favorable Hodgkin's disease. J Clin Oncol. 1988;6:1822–1831. doi: 10.1200/JCO.1988.6.12.1822. [DOI] [PubMed] [Google Scholar]
- 31.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 33.Kramer CY. Extension of multiple range tests to group means with unequal number of replications. Biometrics. 1956;12:307–310. [Google Scholar]
- 34.Cox DR. Regression models and life-tables. J R Stat Soc B. 1972;34:187–220. [Google Scholar]
- 35.Freeman GH, Halton JH. Note on exact treatment of contingency, goodness of fit and other problems of significance. Biometrika. 1951;38:141–149. [PubMed] [Google Scholar]
- 36.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621. [Google Scholar]
- 37.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations of AML1 are common in therapy-related myelodysplasia following therapy with alkylating agents and are significantly associated with deletion or loss of chromosome arm 7q and with subsequent leukemic transformation. Blood. 2004;104:1474–1481. doi: 10.1182/blood-2004-02-0754. [DOI] [PubMed] [Google Scholar]
- 38.Pedersen-Bjergaard J, Andersen MK, Andersen MT, et al. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008;22:240–248. doi: 10.1038/sj.leu.2405078. [DOI] [PubMed] [Google Scholar]
- 39.Levine EG, Bloomfield CD. Leukemias and myelodysplastic syndromes secondary to drug, radiation, and environmental exposure. Semin Oncol. 1992;19:47–84. [PubMed] [Google Scholar]
- 40.Diehl V, Franklin J, Pfreundschuh M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin's disease. N Engl J Med. 2003;348:2386–2395. doi: 10.1056/NEJMoa022473. [DOI] [PubMed] [Google Scholar]
- 41.Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin's lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol. 2009;27:4548–4554. doi: 10.1200/JCO.2008.19.8820. [DOI] [PubMed] [Google Scholar]
- 42.Tucker MA, Coleman CN, Cox RS, et al. Risk of second cancers after treatment for Hodgkin's disease. N Engl J Med. 1988;318:76–81. doi: 10.1056/NEJM198801143180203. [DOI] [PubMed] [Google Scholar]
- 43.Horning SJ, Williams J, Bartlett NL, et al. Assessment of the Stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin's disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol. 2000;18:972–980. doi: 10.1200/JCO.2000.18.5.972. [DOI] [PubMed] [Google Scholar]
- 44.Aversa SM, Trentin C, Sorarù M, et al. Acute promyelocytic leukemia after Stanford V plus radiotherapy for advanced Hodgkin lymphoma. Leuk Lymphoma. 2009;50:1214–1216. doi: 10.1080/10428190902934910. [DOI] [PubMed] [Google Scholar]
- 45.Mistry AR, Felix CA, Whitmarsh RJ, et al. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N Engl J Med. 2005;352:1529–1538. doi: 10.1056/NEJMoa042715. [DOI] [PubMed] [Google Scholar]
- 46.Loeffler M, Brosteanu O, Hasenclever D, et al. Meta-analysis of chemotherapy versus combined modality treatment trials in Hodgkin's disease: International Database on Hodgkin's Disease Overview Study Group. J Clin Oncol. 1998;16:818–829. doi: 10.1200/JCO.1998.16.3.818. [DOI] [PubMed] [Google Scholar]
- 47.Franklin J, Pluetschow A, Paus M, et al. Second malignancy risk associated with treatment of Hodgkin's lymphoma: Meta-analysis of the randomised trials. Ann Oncol. 2006;17:1749–1760. doi: 10.1093/annonc/mdl302. [DOI] [PubMed] [Google Scholar]
- 48.Franklin JG, Paus MD, Pluetschow A, et al. Chemotherapy, radiotherapy and combined modality for Hodgkin's disease, with emphasis on second cancer risk. Cochrane Database Syst Rev. 2005;4:CD003187. doi: 10.1002/14651858.CD003187.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Southwest Oncology Group: Fludeoxyglucose F 18-PET/CT imaging and combination chemotherapy with or without additional chemotherapy and G-CSF in treating patients with stage III or stage IV Hodgkin lymphoma. http://clinicaltrials.gov/show/NCT00822120.