

Abstract
Nucleosomes are the fundamental packing units of the eukaryotic genome. A nucleosome core particle comprises an octameric histone core wrapped around by ~147 bp DNA. Histones and DNA are targets for covalent modifications mediated by various chromatin modification enzymes. These modifications play crucial roles in various gene regulation activities. A group of common hypotheses for the mechanisms of gene regulation involves changes in the structure and structural dynamics of chromatin induced by chromatin modifications. We employed single molecule fluorescence methods to test these hypotheses by monitoring the structure and structural dynamics of nucleosomes before and after histone acetylation and DNA methylation, two of the best-conserved chromatin modifications throughout eukaryotes. Our studies revealed that these modifications induce changes in the structure and structural dynamics of nucleosomes that may contribute directly to the formation of open or repressive chromatin conformation.
Keywords: Nucleosome, Histone acetylation, DNA methylation, Single molecule, FRET, Fluorescence anisotropy
1. Introduction
1.1. Histone+DNA=nucleosome
Core histones wrapped around by DNA make nucleosomes, the fundamental packing units of the eukaryotic genome [1]. Each nucleosome core particle is composed of 145–147 base pair (bp) DNA and two copies of four core histones (H2A, H2B, H3 and H4) that are highly conserved throughout eukaryotes. Nucleosomal DNA bends extensively to form a left-handed superhelix with 1.65 turns around an octameric histone core which comprises a (H3–H4)2 tetramer and two H2A–H2B dimers [2]. Multiple nucleosome core particles joined by 10–90 bp linker DNA, linker histones and some other proteins form chromatin and eventually the chromosome. Electrostatic interactions between core histones and DNA are the main driving force of in vitro nucleosome reconstitution using salt dialysis. In a salt dialysis reconstitution method, the initial 2 M NaCl concentration of a sample solution with core histones and DNA is gradually decreased to 50–150 mM [3]. This method is based on the fact that core histones interact strongly with DNA at a physiological salt concentration while they are stable at a 2 M NaCl concentration without forming contacts with DNA. This reconstitution method suggests that the DNA/histone interactions are stabilized largely by hydrogen bonds and electrostatic interactions between the DNA backbone and the basic residues of histones (R or K) or the helix dipoles of histone-folds [1]. These interactions occur approximately every 10 bp at the minor grooves of nucleosomal DNA [4].
As most of the genes in a cell are wrapped tightly in nucleosomes that are further folded into chromatin, the molecular machines involved in various genomic transactions such as transcription, replication, repair, and recombination need the chromatin and/or nucleosome structure altered in order to access the genetic information. Mechanisms to switch between permissive and repressive states of chromatin conformation by various chromatin modifying and remodeling enzymes have been addressed in detail using various biochemical, molecular biology, epigenetic and single molecule techniques [5–14].
1.2. Core histones
Core histones share a highly conserved tertiary structure despite a low homology in their sequences. The structure represented by the two-fold repetition of a helix/loop/helix configuration with a high degree of helicity (~75%) is the molecular basis for the H2A/H2B and H3/H4 heterodimer formation via a handshake pairing [15]. A four-helix bundle is formed by two major helices and two small helices of two H3 molecules in a nucleosome. In the same way, H2B and H4 molecules also form a four-helix bundle. These helix bundles are the major motives of the octameric structure of a histone core. The residual N-terminal tails that account for ~28% of an octamer core are highly basic due to the abundant lysine and arginine residues and commonly hypothesized to be mostly unstructured [16]. Notably, the N-terminal tails of histones serve as major targets for several posttranslational modifications (PTM) such as acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation that are linked to various gene regulatory activities [17,18]. The N-terminal tails of histones may be in contact with DNA and other parts of histones in intra- and inter-nucleosomal contexts and consequently contribute to the regulation of the structures of nucleosomes and their packaging in chromatin [19–21].
1.3. Nucleosomal DNA sequences suitable for in vitro nucleosome reconstitution
Nucleosome core particles containing various DNA sequences, including a 146 bp fragment derived from the human X-chromosome α-satellite DNA [1] and the Selex 601 sequence [22,23], have been studied with crystallography [24]. A 146 bp fragment derived from a 5s RNA gene has also been used to reconstitute a nucleosome core particle, which was suggested to have two major translational positions [25,26]. The Selex 601 sequence, which was selected from a large random DNA pool by a competition assay, exhibited a high level of af3nity to histones with a strong 10 bp periodicity of the TA dinucleotide sequence that resulted in a highly homogeneous nucleosome position [23].
1.4. Single molecule FRET employed to investigate the structure of nucleosomes
Fluorescence resonance energy transfer (FRET) is non-radiative energy transfer from a donor fluorophore to an acceptor fluorophore [27,28]. The efficiency of FRET is a function of the distance between the two fluorophores. When the acceptor emission can be spectrally well separated from the donor emission, their intensities can be measured separately and the FRET efficiency can be easily calculated with the following equation:
where IA and ID are the fluorescence intensities for the acceptor and the donor, respectively, and ΦA and ΦD are the fluorescence quantum yields of the acceptor and the donor, respectively.
FRET can be modeled as an induced dipole–dipole interaction between a donor and an acceptor. The transfer efficiency as a function of spectral overlap and spatial arrangements of the two fluorophores can be approximated as following.
where r is the distance between the fluorophores, Ro is the Förster distance, κ2 is the orientation factor, ΦD is the donor fluorescence quantum yield, ID is the donor fluorescence emission, and εA is the acceptor absorption.
Combining the above two equations yields
Based on this relationship, one can obtain the distance between the two fluorophores from their fluorescence intensities. When the intensities are measured from individual fluorophores in a time resolved manner, which is referred to as single molecule FRET or smFRET, one can follow the time trajectory of the distance between two fluorophores undergoing FRET.
To calculate the distance between two fluorophores from their FRET efficiency, Ro should be known or experimentally determined. Ro contains many known factors for commercially available fluorophores such as quantum yields and spectral overlap. It also contains the orientation factor κ2 that is 2/3 when the two dipoles are freely rotating. Restriction of fluorophore rotation requires κ2, specific to a given system, to be obtained experimentally or by modeling. For internal comparison of a distance in two different cases, one may avoid κ2 measurement by designing a case-specific approach as we previously demonstrated [12,13]. Utilizing fluorescence anisotropy measurements, fixed fluorophores can be useful to evaluate the structural flexibility of a molecule on a ns timescale or to monitor the changes in the spatial arrangements of two points where the fluorophores are labeled [14].
Single molecule methods are currently being widely employed in studies of dynamic biological macromolecules. Single molecule approaches are particularly useful when the dynamic motion of the entity is complex or the change of interest is too small to be reliably resolved in an ensemble. By employing smFRET measurements we have reported changes, both in structures and structural dynamics of nucleosomes upon histone acetylation and DNA methylation, otherwise buried in noise in ensemble measurements [12–14].
It has long been hypothesized that some chromatin modifications may directly modulate the structure of nucleosomes and chromatin. In particular, histone acetylation and DNA methylation are globally conserved modifications and would have a high chance of affecting the physical properties of nucleosomes such as the structure and structural dynamics. We found that this hypothesis is valid based on our smFRET and fluorescence anisotropy measurements on various nucleosomes. In this review, we will summarize our findings in the effects of histone acetylation and DNA methylation on the structure and dynamics of nucleosomes addressed from single molecule approaches [12–14].
2. Effects of histone acetylation on the structure of nucleosomes and internucleosomal interactions
Core histone tails contain lysine residues that serve as targets for various histone acetyltransferases, which transfer an acetyl group of acetyl-coenzyme A (Ac-CoA) to the ε-amine group of a lysine residue [29,30]. A long-standing hypothesis for histone acetylation affecting chromatin structure is based on charge neutralization of the lysine residues upon acetylation. The charge neutralization would reduce the affinity of lysine to DNA, resulting in an elevated level of DNA accessibility. Early studies reported that hyperacetylation of nucleosomes increases the susceptibility of nucleosomal DNA to DNase I digestion and enhances thermal denaturation of nucleosomes, supporting this hypothesis [31]. A large number of components in transcriptional regulatory proteins such as TAF130/250, p300/CBP, ACTR, and SRC-1, possess intrinsic histone acetyltransferase activity [32]. Therefore, in addition to the recruitment of chromatin modification enzymes, histone acetylation by these proteins may also contribute to histone eviction or partial disassembly of nucleosomes to open up the tightly wrapped genes out of nucleosomes for transcription. This hypothesis is particularly important to address changes happening in promoter nucleosomes as RNA polymerases can pass through nucleosomes in open reading frames without dissociating histones [33].
2.1. Effects of histone acetylation on the structure of nucleosomes
Structural changes of nucleosomes upon histone acetylation have been investigated since histone acetylation was linked to elevated transcriptional activity in eukaryotic cells in 1964 [32,34]. Discovered effects of histone acetylation on nucleosome structure, albeit subtle, include decrease in the linking number of nucleosomal DNA [35], transient unwrapping of DNA from the edge of the nucleosome, and altered hydrodynamic behavior [31]. Based on smFRET and fluorescence anisotropy measurements, we characterized the changes in the structure of nucleosomes upon histone acetylation by a functional sub-complex of a histone acetyltransferase called Nucleosome Acetyltransferase of Histone H4 (NuA4), Piccolo NuA4, which acetylates mainly H4 and H2A tails [13,36,37].
In our experiments, several oligonucleotide fragments, two of which were labeled with a FRET pair (Cy3 and Cy5), were ligated to construct the 147 bp 601 nucleosomal DNA with an additional surface linker at one end (Fig. 1) [13]. The linker was 10 nucleotides in length and labeled with a biotin at one end for surface immobilization. The fluorophores were labeled along the backbone of the DNA and two ends of a fluorophore molecule are anchored to the backbone. According to our routine gel shift assay, this labeling does not change the hydrodynamic behavior of nucleosomes, suggesting that the nucleosomal DNA structure is not altered severely. The two dyes were separated by 65–83 bp to yield 0.6–0.7 FRET efficiency upon nucleosome formation as estimated from a crystal structure (Fig. 2). Nucleosomes were assembled with recombinant Xenopus laevis histones using the yeast nucleosome assembly protein 1 (yNAP1) histone chaperone. yNAP1 is an acidic protein that binds histones with high affinity and is associated with histone trafficking, nucleosome assembly and disassembly [38–40]. yNAP1 is commonly used in nucleosome assembly under physiological ionic conditions [41]. It assembles nucleosomes by depositing a (H3–H4)2 tetramer on DNA to make a tetrasome and subsequent transfer of two H2A–H2B dimers [42]. Reconstituted 601 nucleosomes were analyzed using native-PAGE to display one major band, indicating no significant positioning heterogeneity. The homogeneous nucleosome positioning is a property of the 601 sequence well-characterized by micrococcal nuclease digestion assays [22,43]. Nucleosomes were immobilized on the surface of a quartz microscope slide via biotin–streptavidin conjugation for the smFRET measurements. Fluorescence intensities of the two fluorophores from individual nucleosomes were recorded with a sensitive charge coupled device (CCD) camera in a time resolved manner with a typical temporal resolution of 15–35 ms, generating a movie with a 28–67/s frame rate (A note on the temporal resolution: with 15 ms resolution, some structural excursions to a state with a lifetime on the timescale of a few ms can easily be detected because this type of dynamics typically follows exponential decay population statistics. For instance, 5% of the total population of a system in a state with 5 ms lifetime has the dwell time longer than 15 ms in the state assuming a single exponentially decaying population density along time). One movie contains information on the time-resolved structural dynamics of 300–500 nucleosomes that were spatially resoluble (i.e. 300–500 separated fluorescent dots on the movie screen). FRET efficiencies at each time point were calculated as described in the introduction and plotted along time to show the time trace of the FRET efficiency (Fig. 1B). No structural dynamics were noticeable in the FRET traces of nucleosomes either acetylated or not, indicating stable structures of nucleosomes in the timescale of a few milliseconds or longer. The average FRET efficiency in each case was obtained by fitting the FRET efficiency histogram with a Gaussian distribution function (Fig. 2). It was revealed that the average FRET efficiency changes when nucleosomes were incubated with Piccolo NuA4 in the presence of Ac-CoA. The FRET efficiency decreases upon the acetylation in case of the 601A nucleosomes while it increases in case of the 601B nucleosomes. These FRET changes indicated that nucleosomal DNA unwraps upon histone acetylation by Piccolo NuA4.
Fig. 1.

Schematics of our single molecule nucleosome experimental system. A. The 147 bp nucleosomal DNA used in the experiments comprises a ~60 bp (H3–H4)2 tetramer binding site (blue) and two ~30 bp binding sites for H2A/H2B dimers (orange) near the termini. One (H3–H4)2 tetramer and two H2A–H2B dimers are shown in the same color in a crystal structure (PDB: 3MVD) [12,13]. Methylation sites are marked with black spheres [12]. The list of acetylation sites is taken from a review by Turner [65]. B. Nucleosomes labeled with fluorophore(s) are immobilized on a surface via biotin–streptavidin conjugation [12,13]. Fluorescence signals from single Cy3 and Cy5 fluorophores were imaged on a CCD camera. The fluorescence intensities recorded in a time resolved manner (every 210 ms) were converted to a FRET efficiency time trace as described in the text.
Fig. 2.
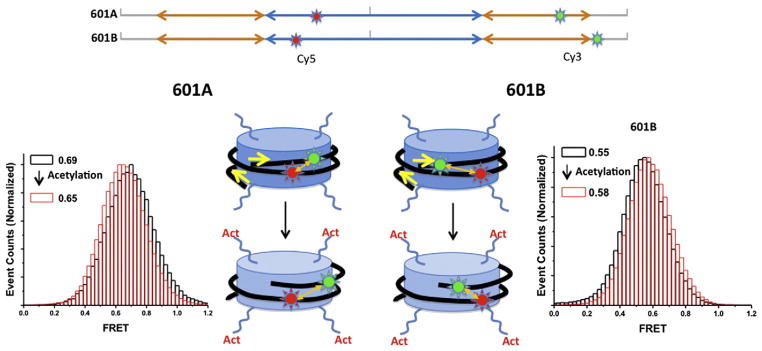
Effects of histone acetylation on the structure of nucleosomes. 601A and 601B nucleosomes labeled with Cy3 and Cy5 displayed FRET efficiencies of 0.69 and 0.55, respectively (black in histogram) [13]. Signal integration time is 35 ms. Upon acetylation (red in histogram), the FRET efficiency of 601A nucleosome was decreased whereas the FRET efficiency of 601B nucleosomes was increased [13].
Nucleosomal DNA is overly twisted to form a superhelix around a histone core. Therefore, if DNA unwraps from the histone core, the twistedness [22] of the DNA must be altered and consequently its topology must change. As the fluorophores in the nucleosomes are labeled along the phosphate backbone of the DNA, they maintain fixed dipole orientations. A topology change of DNA, therefore, must result in altered dipole orientations of the dyes. The altered dipole orientations of a FRET pair can be verified with fluorescence anisotropy of the acceptor excited via FRET from the donor [12,13]. In order to measure the anisotropy change due to the altered dipole orientation, fluorescence anisotropy decay due to tumbling of nucleosomes must be compensated to the measured anisotropy by using the following equation [12]:
where rrot + dipole is the measured anisotropy of acceptor emission via FRET, rrot,D and rrot,A are respectively the average emission anisotropy of the donor and acceptor with the direct excitations, rdipole is the an-isotropy containing the decay only due to the skewed dipole between the donor emission and acceptor excitation, ϕ is the fluorescence quantum efficiency of the donor, and E is the FRET efficiency. When the FRET efficiency and anisotropy are reasonably high and the quantum efficiency is low, this equation can be approximated as the last term in the equation. The rdipole can be related to the angle between the two fluorophores as follows [44]:
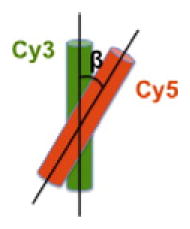
A change in β would also result in a change in FRET efficiency. By decoupling the β change from the distance change, we showed that both topology and distance are changed upon histone acetylation [13]. This result supports that DNA unwrapping is accompanied by a topology change, which is likely a signature of DNA untwisting. This result is in line with the acetylation-induced writhe of DNA resulting in the release of negative supercoils of nucleosomal DNA [35,45]. The unwrapping and unwinding of DNA may result in an enhanced accessibility of DNA. As opening of the DNA regions in contact with H2A-H2B is commonly hypothesized to be the first step of nucleosome disassembly [46], destabilization of a nucleosome upon acetylation suggested by our observation may facilitate disassembly of a nucleosome. Either via increased accessibility of DNA within an intact nucleosomal context or facilitated disassembly of nucleosomes, histone acetylation may contribute to gene activation by altering the structure of nucleosomes.
2.2. Effects of histone acetylation on internucleosomal interactions
Previous studies [1,10,47] suggest that the basic N-terminal tails of histone H4 (residues 14–19) interact with acidic patches of H2A in an internucleosomal manner through multiple hydrogen bonds and salt bridges, raising a possibility that acetylation of H4 tails will influence internucleosomal interactions and consequently the structure of chromatin. In a model for 30 nm chromatin fiber based on the crystal structure of a tetranucleosome, the H4 tail in one nucleosome and the H2A–H2B dimer in a neighboring nucleosome are positioned in close proximity to each other [48]. In the tetranucleosome used in the crystallographic study (4×601 nucleosomes joined by 20 bp linker DNA), the internucleosomal histone-histone interactions are affected by the spatial constraints imposed by the linker DNA [48]. In order to study the intrinsic internucleosomal histone–histone interactions free from the effect of linker DNA, we monitored the interactions with freely diffusing nucleosomes in solution at a single molecule level. This approach is a unique benefit of the single molecule nucleosome system that we developed. In our system, nucleosomes are attached on the surface of a microscope slide that is functionalized with poly-ethyleneglycol. This system provides an extremely efficient means to monitor individual nucleosomes and their interactions with solution-born entities (e.g. diffusing nucleosomes and nucleosome binding proteins) in a time resolved manner, thereby making complex dynamics and subtle changes in the structures easily resoluble.
In our setup, a set of nucleosomes labeled with a FRET donor (Cy3) were immobilized on a surface of a sample chamber and the other set of nucleosomes labeled with a FRET acceptor (Cy5) were injected to the chamber (Fig. 1B) [13]. FRET signal arises when a freely diffusing Cy5 labeled nucleosome forms a dinucleosome with a surface immobilized Cy3 labeled nucleosome (Fig. 3B). Various FRET states were identified, indicating the existence of multiple dinucleosomal states (Fig. 3C). The observed dinucleosomal states were mostly transient, which were categorized as short-lived (less than a hundred milliseconds) and long-lived (several hundred milliseconds or longer) states (Fig. 3C). Upon histone acetylation, the efficiency of dinucleosome formation was considerably reduced (26.5%→ 6.1%) and the lifetime of the long-lived dinucleosomes was significantly shortened (1.12 s→ 0.58 s). In addition, the FRET efficiency of the long-lived dinucleosomal states was decreased. These results suggest that the internucleosomal distance is increased and consequently the internucleosomal interface becomes more accessible upon histone acetylation. The FRET efficiencies are also Mg2+ ion-dependent, which strongly supports that the internucleosomal interactions we observed are relevant to the Mg2+-dependent higher order chromatin folding [49].
Fig. 3.

Effects of histone acetylation on internucleosomal interactions. A. Schematic representation of fluorophore positions on a 601 DNA fragment to study nucleosome interactions [13]. Each nucleosome is assembled with a fluorophore, either Cy3 or Cy5. Cy3 labeled 601C nucleosomes were immobilized on a surface and Cy5 labeled 601D nucleosomes or 601E nucleosomes were injected to the sample chamber [13]. B. A typical FRET time trace revealing the spontaneous formation of dinucleosomes between 601C and 601D nucleosomes [13]. C. Lifetime histograms of the dinucleosomal states were constructed, which revealed a long-lived and a short-lived population. The long-lived dinucleosomal states were collected in the FRET time traces and histograms of their FRET efficiencies were constructed to reveal that there is a overall decrease in the FRET efficiency upon the acetylation [13]. D. Schematic representation of the most compact dinucleosomal state modeled with the crystal structure of a nucleosome core particle (3MVD) [13].
Based on the FRET efficiencies obtained from two dinucleosomes with two different FRET pair locations, we estimated the structure of the most stable dinucleosomal state (Fig. 3D) [13]. We found that the two nucleosomes in the dinucleosome are rotated by 34° with respect to each other and separated by ~2.0 nm. In the tetranucleosome crystal structure, two nucleosomes were separated by ~1.76 nm, indicating a comparable extent of compaction between nucleosomes [48]. This agreement suggests that intrinsic histone-histone interactions play a major role in the nucleosome packaging in chromatin. Our estimate and the suggested model based on the crystal structure puts histones H4 and H2A in close proximity, confirming the suggestion based on biochemical studies that the interaction between H4 and H2A helices may play a significant role in nucleosome packaging in chromatin [10,47]. Among the four histone acetylation sites on H4N-terminal tail, H4K16 is of particular interest as it was found solely responsible for internucleosomal and inter-array compaction [11,50]. It will be important in the future to acetylate specifically at each of the four sites to identify the roles for each acetylation site in the internucleosomal and inter-array interactions.
Based on these results, we have suggested that histone acetylation contributes to the activation of genes both by inducing a more open structure of nucleosomes and by disturbing higher order folding of chromatin [29].
3. Effects of DNA methylation on the structure of nucleosomes
DNA methylation is associated with repressive chromatin states. It is carried out by a DNA methyltransferase which transfers a methyl group from S-adenosyl methionine (SAM) to the C5 position of cytosine in a CpG dinucleotide context in most cases [51]. Approximately 4% of the genomic DNA is hypermethylated primarily in the form of 5-methylcytosines in CpG dinucleotides [52]. CpG dinucleotides in vertebrate DNA appear at a frequency lower than the statistically expected value [53]. On the other hand, stretches of vertebrate DNA with close to or higher than the expected frequency of CpG dinucleotides, so called “CpG islands” have also been identified [54,55]. CpG islands are typically defined as a 200-bp or longer stretch of DNA with a C+G content of 50% or higher and the ratio of observed CpG/expected CpG in excess of 0.6 [53]. CpG islands are often associated with the 5′ ends near the promoters for housekeeping genes and many tissue-specific genes, and sometimes with the 3′ ends of some tissue-specific genes [53]. Most promoter-associated CpG islands in normal somatic cells and germ-line tissues remain hypomethylated, permitting gene expression. In contrast, CpG islands in the promoters of tumor-suppressors in most cancer cells are hypermethylated, likely contributing to carcinogenesis [56–58].
DNA methylation could contribute to the formation of repressive chromatin in two ways. The methyl groups could alter the physical properties of DNA, which would affect the structure of nucleosomes and contribute to the formation of repressive chromatin. In the other mechanism, methyl-binding proteins such as transcriptional repressors that recruit histone deacetylases maintain histones deacetylated and turn off transcription. These two hypotheses are not mutually exclusive. In order to test the first hypothesis, we monitored the changes in the structure and structural dynamics of nucleosomes upon CpG methylation based on smFRET and anisotropy measurements.
3.1. Effects of CpG methylation on the structure and structural dynamics of the terminal regions of nucleosomal DNA
We used a nucleosome construct similar to those described in the acetylation section to study the effects of CpG methylation on the structure and structural dynamics of the termini of nucleosomal DNA [14]. Nucleosomes were reconstituted with Xenopus laevis histones and a fragment of 5S rDNA from sea urchin using yNAP1. The FRET pair was labeled at the termini of nucleosomal DNA in order to monitor changes in the termini dynamics upon CpG methylation. The DNA was methylated with a methyltransferase enzyme. The assembled nucleosomes, unlike the 601 nucleosomes, displayed two major FRET efficiencies (Fig. 4A and B), indicating two major groups of nucleosome positions on the DNA as suggested by Luger et al. [25]. In order to avoid this positioning heterogeneity, we started using the 601 sequence for later studies. No noticeable termini dynamics on the timescale of a few milliseconds or longer was observed from either of the two FRET populations. Upon methylation, short excursions to a very high FRET state were observed from both populations (Fig. 4C and D). These short excursions could mean that the termini wrapped around the histone core tightly and transiently, or that the termini freely diffused and transiently encountered each other. In order to verify which possibility was the case, we measured the fluorescence anisotropy of the acceptor excited via FRET from the donor. As the fluorophores are labeled at the fiexible termini of nucleosomal DNA, their structural fluctuations in the timescale of the fluorescence decay must contribute to the anisotropy decay. If the termini wrapped more tightly upon methylation, the termini dynamics will be significantly reduced in the high FRET state and consequently the anisotropy will be maintained at a high level. Being able to obtain the anisotropy at each time point of the measurements with our single molecule setup, we found that the high FRET state has a very high anisotropy level, confirming that DNA termini are tightly wrapped around the histone core in the high FRET states. Therefore, on average, CpG methylation induces compaction and rigidity in the structure of nucleosomes. Other studies also support our findings on the rigidified DNA upon CpG methylation [59,60]. One such example was the reduced flexibility of the phosphate-sugar backbone in the DNA dodecamer containing C9pG10 methylation. In another study, bulky methyl groups were suggested to restrict the conformational space of a methylated 32-mer cAMP responsive element, resulting in an elevated level of stiffness.
Fig. 4.

Effects of CpG methylation on the structure and structural dynamics of the terminal regions of nucleosomal DNA.5S rDNA nucleosomes display two unevenly distributed FRET states, a 0.29 FRET state (A) and a 0.4 FRET state (B) [14]. Signal integration time is 15 ms. After incubation with CpG methyltransferase, the population of nucleosome showing excursions to a high 0.74 FRET state was increased from 0.4% to 16% [14]. Examples of smFRET time trajectories that displayed excursions to a high 0.74 FRET state from 0.29 and 0.4 FRET states are shown in (C) and (D), respectively.
These results provide a physical basis for how CpG methylation may contribute to the formation of repressive chromatin structure by altering nucleosome dynamics. In addition to acting as “molecular handles” recognized by methyl-CpG binding proteins, our results suggest a novel role for CpG methylation in regulating interactions between histones and DNA by altering structural dynamics of nucleosomal DNA. This may also have important consequences on folding of nucleosome arrays as well as interactions with chromatin-associated proteins.
3.2. Effects of CpG methylation on the structure of the internal regions of nucleosomal DNA
The compaction induced upon CpG methylation must alter the histone-DNA interactions and consequently the structure of the internal regions of nucleosomal DNA. We used 601 nucleosomes (Xenopus laevis histones and the 601 sequence) labeled with a FRET pair in order to test this hypothesis [12]. The same experimental setup and analysis scheme as introduced in the acetylation section were employed. According to the smFRET time traces, no dynamics in the timescale of a few milliseconds or longer were observed. The structure of the internal regions of nucleosomal DNA showed significant changes upon CpG methylation (Fig. 5). The direction of the changes indicated that nucleosomal DNA tightly wrapped the core histones upon CpG methylation.
Fig. 5.

Effects of CpG methylation on the internal regions of nucleosomal DNA. 601+39 nucleosomes and 601+29 nucleosomes displayed FRET changes indicating tight wrapping of DNA around the histones upon CpG methylation [12]. 601+39 and 601+29 nucleosomes are labeled with Cy3 (−38) and Cy5 (+39 or +29) on DNA [12]. Signal integration time is 25 ms. FRET efficiencies of 601+39 and 601+29 were 0.84 and 0.66 (grey in histogram), respectively, which changed to 0.71 and 0.76 (black in histogram), respectively, upon methylation [12].
This result is in line with the Monte Carlo simulation study, which suggested an increased helical repeat length of DNA with methylated CpGs as a result of DNA untwisting [61]. Increased helical repeat length will require an elevated level of core histone content to package a given length of DNA, which would result in compaction of chromatin structure. In order to test this hypothesis, we employed the same topology change assay as described in the acetylation section [12]. Based on the topology change we observed upon methylation, we suggested that the tight wrapping is due to the untwisting of DNA.
According to these findings, CpG methylation in the CpG islands may directly contribute to the formation of repressive chromatin states. CpG islands are often concentrated in the vicinity of promoter regions where transcription is initiated [62]. When CpG dinucleotides are within or close to nucleosomal regions, the DNA topology change induced upon CpG methylation would result in changes in the rotational and/or translational settings of the nucleosomes. As subtle changes in the rotational or translational settings of nucleosomes in promoter regions can dramatically modulate the efficiency of transcription initiation, CpG methylation in these regions may directly impact on transcription initiation. To support this hypothesis, it has been reported that small changes in the translational or rotational settings of nucleosomes inhibit the access of a TATA box binding protein to its target site, preventing transcription [63,64].
These findings suggest that CpG methylation, in addition to its role as a molecular handle for CpG binding proteins, contributes directly to the formation of repressive chromatin structures by altering the interactions between histones and DNA.
4. Conclusions
Recent advancement in identifying chromatin-binding proteins has shed light on how genes are regulated by the catalytic activities of these proteins and their interactions with chromatin. However, we still lack a basic biophysical understanding of how these proteins contribute to the regulation of the structure and structural dynamics of chromatin, which are another critical aspect of chromatin dynamics in cells. The new insights brought by recent studies in our lab demonstrate the richness of information that single molecule methods can contribute to the field of chromatin biology.
Acknowledgments
Our research was funded by an NIH grant (GM097286), a Searle Scholar award and a Henry and Camille Dreyfus New Faculty Award to T.L.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Richmond TJ. DNA binding within the nucleosome core. Curr Opin Struct Biol. 1998;8:33–40. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- 3.Wilhelm FX, Wilhelm ML, Erard M, Daune MP. Reconstitution of chromatin: assembly of the nucleosome. Nucleic Acids Res. 1978;5:505–521. doi: 10.1093/nar/5.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buning R, Van Noort J. Single-pair FRET experiments on nucleosome conformational dynamics. Biochimie. 2010;92:1729–1740. doi: 10.1016/j.biochi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27:389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 7.Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 8.Cairns BR. Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 10.Kan PY, Caterino TL, Hayes JJ. The H4 tail domain participates in intra-and internucleosome interactions with protein and DNA during folding and oligomerization of nucleosome arrays. Mol Cell Biol. 2009;29:538. doi: 10.1128/MCB.01343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 12.Lee JY, Lee TH. Effects of DNA methylation on the structure of nucleosomes. J Am Chem Soc. 2012;134:173–175. doi: 10.1021/ja210273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JY, Wei S, Lee TH. Effects of histone acetylation by Piccolo NuA4 on the structure of a nucleosome and the interactions between two nucleosomes. J Biol Chem. 2011;286:11099. doi: 10.1074/jbc.M110.192047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee TH. DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arents G, Moudrianakis EN. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci. 1995;92:11170. doi: 10.1073/pnas.92.24.11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luger K, Richmond TJ. The histone tails of the nucleosome. Curr Opin Genet Dev. 1998;8:140–146. doi: 10.1016/s0959-437x(98)80134-2. [DOI] [PubMed] [Google Scholar]
- 17.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part I: Covalent histone modifications. Trends Mol Med. 2007;13:363–372. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Arya G, Schlick T. A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments?? J Phys Chem A. 2009;113:4045–4059. doi: 10.1021/jp810375d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertin A, Renouard M, Pedersen JS, Livolant F, Durand D. H3 and H4 histone tails play a central role in the interactions of recombinant NCPs. Biophys J. 2007;92:2633–2645. doi: 10.1529/biophysj.106.093815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 22.Widom J. Role of DNA sequence in nucleosome stability and dynamics. Q Rev Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- 23.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning1. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 24.Makde RD, England JR, Yennawar HP, Tan S. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 2010;467:562–566. doi: 10.1038/nature09321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luger K. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev. 2003;13:127–135. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 26.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 27.Förster T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann Phys. 1948;437:55–75. [Google Scholar]
- 28.Stryer L, Haugland RP. Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci U S A. 1967;58:719. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 30.Hansen JC, Nyborg JK, Luger K, Stargell LA. Histone chaperones, histone acetylation, and the fluidity of the chromogenome. J Cell Physiol. 2010;224:289–299. doi: 10.1002/jcp.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ausio J, Van Holde KE. Histone hyperacetylation: its effects on nucleosome conformation and stability. Biochemistry. 1986;25:1421–1428. doi: 10.1021/bi00354a035. [DOI] [PubMed] [Google Scholar]
- 32.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 33.Kulaeva OI, Gaykalova DA, Pestov NA, Golovastov VV, Vassylyev DG, Artsimovitch I, Studitsky VM. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat Struct Mol Biol. 2009;16:1272–1278. doi: 10.1038/nsmb.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Norton VG, Marvin KW, Yau P, Bradbury EM. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J Biol Chem. 1990;265:19848. [PubMed] [Google Scholar]
- 36.Berndsen CE, Selleck W, McBryant SJ, Hansen JC, Tan S, Denu JM. Nucleosome recognition by the Piccolo NuA4 histone acetyltransferase complex. Biochemistry. 2007;46:2091–2099. doi: 10.1021/bi602366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selleck W, Fortin I, Sermwittayawong D, Cote J, Tan S. The Saccharomyces cerevisiae Piccolo NuA4 histone acetyltransferase complex requires the Enhancer of Polycomb A domain and chromodomain to acetylate nucleosomes. Mol Cell Biol. 2005;25:5535. doi: 10.1128/MCB.25.13.5535-5542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell. 2010;37:834–842. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrews AJ, Downing G, Brown K, Park YJ, Luger K. A thermodynamic model for Nap1–histone interactions. J Biol Chem. 2008;283:32412–32418. doi: 10.1074/jbc.M805918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park YJ, Chodaparambil JV, Bao Y, McBryant SJ, Luger K. Nucleosome assembly protein 1 exchanges histone H2A–H2B dimers and assists nucleosome sliding. J Biol Chem. 2005;280:1817. doi: 10.1074/jbc.M411347200. [DOI] [PubMed] [Google Scholar]
- 41.Fujii-Nakata T, Ishimi Y, Okuda A, Kikuchi A. Functional analysis of nucleosome assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J Biol Chem. 1992;267:20980–20986. [PubMed] [Google Scholar]
- 42.Bohm V, Hieb AR, Andrews AJ, Gansen A, Rocker A, Toth K, Luger K, Langowski J. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 2011;39:3093. doi: 10.1093/nar/gkq1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rochman M, Postnikov Y, Correll S, Malicet C, Wincovitch S, Karpova TS, McNally JG, Wu X, Bubunenko NA, Grigoryev S, Bustin M. The interaction of NSBP1/HMGN5 with nucleosomes in euchromatin counteracts linker histone-mediated chromatin compaction and modulates transcription. Mol Cell. 2009;35:642–656. doi: 10.1016/j.molcel.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lakowicz JR, Masters BR. Principles of fluorescence spectroscopy. J Biomed Opt. 2008;13:029901. [Google Scholar]
- 45.Norton VG, Imai BS, Yau P, Bradbury EM. Histone acetylation reduces nucleosome core particle linking number change. Cell. 1989;57:449–457. doi: 10.1016/0092-8674(89)90920-3. [DOI] [PubMed] [Google Scholar]
- 46.Böhm V, Hieb AR, Andrews AJ, Gansen A, Rocker A, Tóth K, Luger K, Langowski J. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 2011;39:3093. doi: 10.1093/nar/gkq1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinha D, Shogren-Knaak MA. Role of direct interactions between the histone H4 tail and the H2A core in long range nucleosome contacts. J Biol Chem. 2010;285:16572. doi: 10.1074/jbc.M109.091298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 49.Hansen JC. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- 50.Robinson PJJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol. 2008;381:816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 52.Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2:S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 53.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 54.Tykocinski ML, Max EE. CG dinucleotide clusters in MHC genes and in 5?? demethylated genes. Nucleic Acids Res. 1984;12:4385–4396. doi: 10.1093/nar/12.10.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 56.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 57.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 58.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 59.Geahigan KB, Meints GA, Hatcher ME, Orban J, Drobny GP. The dynamic impact of CpG methylation in DNA. Biochemistry. 2000;39:4939–4946. doi: 10.1021/bi9917636. [DOI] [PubMed] [Google Scholar]
- 60.Derreumaux S, Chaoui M, Tevanian G, Fermandjian S. Impact of CpG methylation on structure, dynamics and solvation of cAMP DNA responsive element. Nucleic Acids Res. 2001;29:2314–2326. doi: 10.1093/nar/29.11.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nathan D, Crothers DM. Bending and flexibility of methylated and unmethylated EcoRI DNA1. J Mol Biol. 2002;316:7–17. doi: 10.1006/jmbi.2001.5247. [DOI] [PubMed] [Google Scholar]
- 62.List AF. New approaches to the treatment of myelodysplasia. Oncologist. 2002;7:39–49. doi: 10.1634/theoncologist.7-suppl_1-39. [DOI] [PubMed] [Google Scholar]
- 63.Imbalzano AN, Kwon H, Green MR, Kingston RE. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature. 1994;370:481–485. doi: 10.1038/370481a0. [DOI] [PubMed] [Google Scholar]
- 64.Martinez-Campa C, Politis P, Moreau JL, Kent N, Goodall J, Mellor J, Goding CR. Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail and the bromodomain factor Bdf1. Mol Cell. 2004;15:69–81. doi: 10.1016/j.molcel.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 65.Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]