

Abstract
The human pathogen Giardia lamblia is an anaerobic protozoan parasite that causes giardiasis, one of the most common diarrheal diseases worldwide. Although several drugs are available for the treatment of giardisis, resistance to these drugs has been reported and is likely to increase. The Giardia carbamate kinase (glCK) plays an essential role in Giardia metabolism and has no homologs in humans, making it an attractive candidate for anti-Giardia drug development. We have developed a luminescent enzyme coupled assay to measure the activity of glCK by quantitating the amount of ATP produced by the enzyme. This assay is homogeneous and has been miniaturized into a 1536-well plate format. A pilot screen against 4,096 known compounds using this assay yielded a signal-to-basal ratio of 11.5 fold and Z’ factor of 0.8 with a hit rate of 0.9 % of inhibitors of glCK. Therefore, this Giardia lamblia carbamate kinase assay is useful for high throughput screening of large compound collection for identification of the inhibitors for drug development.
Keywords: Carbamate kinase, Giardia, high throughput screening, assay development.
INTRODUCTION
The protozoan parasite Giardia lamblia is a human pathogen with worldwide health impact of 280 million annual cases and 10000 deaths [1, 2]. The parasite causes the diarrheal disease giardiasis, which can lead to acute and chronic diarrhea, malnutrition and failure to thrive. The standard of care for giardiasis is either metronidazole or tinidazole, with single course cure rates of 80–95%, while other drugs such as paromomycin, albendazole and nitazoxanide are used to a lesser extent with similar and/or lower success rates [3]. Current drugs are predominantly of the nitroimidazole or benzimidazole classes and are susceptible to mechanisms such as down-regulation of pyruvate:ferredoxin oxidoreductase (PFOR) and mutations of β-tubulin [1]. Although these drugs are generally effective, treatment failures and drug resistant strains have been increasingly reported [2, 4, 5], highlighting the need for novel drug targets.
The three-step arginine dihydrolase pathway, involving arginine deiminase, ornithine transcarbamoylase and carbamate kinase (CK, EC 2.7.2.2), is used as a major source of energy generation by a number of anaerobic prokaryotes [6, 7] and some parasitic protozoa such as trichomonads [8] and G. lamblia [9, 10]. In G. lamblia, the arginine dihydrolase pathway is not only a major source for energy production, but also plays a role in host colonization and immune system evasion [11, 12]. Of the three enzymes involved in this pathway, the G. lamblia CK (glCK) was found to be the most active, with a maximal activity in cell free extracts that was 5- to 10-fold greater than those of arginine deiminase and ornithine transcarbamoylase [10]. In addition, glCK has been shown to be essential for the viability of G. lamblia WB trophozoites [13]. The essentiality of glCK and the lack of the arginine dihydrolase pathway in higher eukaryotes place glCK as a potential anti-giadial target.
CK catalyzes the reversible conversion of carbamoyl phosphate and ADP to ATP, ammonia and CO2 (Fig. 1). Several assays are available for both the forward and reverse CK reaction, but are limited by throughput. For the forward reaction, CO2, one of three products, can be measured directly as a gas released with limited assay throughput as a specialized apparatus is needed for the measurement [14]. Measurement of another product ATP has been achieved by coupling the CK reaction to hexokinase and glucose-6-phosphate dehydrogenase to produce NADPH, which is then detected by 340 nm absorbance [13, 15]. For the reverse CK reaction, two enzyme coupled assays are available: (1) the CP product can be assayed by coupling to ornithine transcarbamylase to produce citrulline, which is monitored colorimetrically [15], and (2) the ADP product can be assayed by coupling to pyruvate kinase and lactate dehydrogenase to oxidize NADH, which is monitored by 340 nm absorbance [13]. However, absorbance and colorimetric assays typically have limited sensitivity and robustness and are not ideal for HTS in miniaturized plate formats. We have developed a new luminescence-based assay for the forward CK reaction that detects the production of ATP. This assay has been successfully miniaturized to 1536-well format and validated in a pilot screen of 4096 small molecule compounds.
Fig. (1).

Assay Principles.
MATERIALS AND METHODS
Reagents
Adenosine 5’-diphosphate potassium salt (ADP, EMD Millipore, Darmstadt, Germany), Adenosine 5’-triphosphate disodium salt (ATP) and carbamoyl phosphate dilithium salt (CP, Sigma-Aldrich, St. Louis, MO) were purchased as powder samples and dissolved in deionized water. ATPLiteTM assay kit (PerkinElmer, Waltham, MA) for quantitating amount of ATP was reconstituted according to vendor directions. Recombinantly expressed and purified glCK was prepared as previously described [13]. All micro-titer plates used in this study were purchased from Greiner Bio One (Monroe, NC).
Small Molecule Libraries and Compound Management
The library of pharmacologically active compounds (LOPAC1280) consists of a collection of 1,280 small molecules with known biological activities. The LOPAC1280 library has been extensively used for HTS assay validations [16, 17] and was purchased from Sigma-Aldrich. The NIH Chemical Genomics Center (NCGC) Pharmaceutical collection (NPC) was constructed internally through a combined effort of compound purchasing and custom synthesis [18]. At the time of screening, the NPC library consisted of 2816 small molecule compounds, 52% of which were drugs approved for human or veterinary use by the United States Food and Drug Administration (FDA), 22% were drugs approved in Europe, Canada or Japan, and the remaining 25% were compounds that entered clinical trials or were research compounds commonly used in biomedical research. Compounds from both libraries were obtained as powder samples and dissolved in DMSO as 10 mM stock solutions, except for several hundred compounds from the NPC library that were prepared as 4.47 mM stock solutions due to solubility limitations. All library compounds were serially diluted in DMSO via 1:5 inter-plate titrations for primary screens, then formatted to 1536-well compound plates using an Evolution P3 system (PerkinElmer, Waltham, MA) [19]. After formatting, compound plates were stored desiccated at room temperature for as long as 6 months when in use, or heat sealed and stored at -80 oC for long-term storage. Compound cherry-picking for hit confirmation was done as follows: 10 mM compound solutions were thawed from storage, diluted in DMSO as 12-point 1:3 intra-plate titrations, and then formatted to 1536-well compound plates.
Luminescence-Based Carbamate Kinase Assay in 384-Well Format
All assay optimizations were carried out in white, 384-well, low volume, white, solid bottom, medium binding plates (Greiner Bio-one, Monroe, NC). Unless otherwise noted, 4 µl/well of 1 nM glCK enzyme in assay buffer (20 mM Tris pH 8, 30 mM MgCl2, 0.1% BSA, 0.05% Tween 20) was dispensed with a BioRAPTR FRD Microfluidic Workstation (Beckman Coulter, Brea, CA). Subsequently, the reaction was started by simultaneously dispensing 2 µl/well of substrate mix (600 µM ADP and 600 µM carbamate phosphate in assay buffer) and 2 µl/well of the detection reagent (ATPLiteTM) with the FRD Workstation. The reaction was allowed to proceed at room temperature for 20 min, and then the signal was read on a ViewLux CCD-based imaging plate reader (PerkinElmer, Waltham, MA) using a clear filter.
The steady-state kinetics were determined with 2.5 nM glCK, 2 mM of the saturating substrate and the variable substrate at concentrations of 0.076, 0.23, 0.69, 2.1, 6.2, 18.5, 55.6, 167 µM. The reaction signals were read at every minute for 5 min to calculate the initial velocities (V0). The Michaelis constant (Km) and Kcat values were calculated from the V0 values as a function of the concentration of the varied substrate, [S] by fitting with GraphPad Prism to the equation V0 = Vmax [S] / (Km + [S]) to obtain the maximum velocity (Vmax) and the Km. Kcat was calculated from the ratio of Vmax and the pMol enzyme used in each reaction.
Luminescence-Based Carbamate Kinase Assay in 1536-Well Format
The glCK assay was carried out in 1536-well, medium-bind, white, solid bottom plates (Greiner Bio-one, Monroe, NC) as listed in Table 1. Briefly, 2 µl/well of 1 nM glCK enzyme in assay buffer was dispensed with the FRD Workstation. Compounds dissolved in DMSO were added from a source plate to an assay plate at a volume of 23 nl/well by an automated pin-tool station (Wako-Kalypsys, San Diego, CA). The enzyme was allowed to equilibrate with compounds for 15 min at room temperature. Subsequently, the reaction was started with the simultaneous dispensing of 1 µl/well of substrate mix (600 µM ADP and 600 µM carbamate phosphate in assay buffer) and 1 µl/well of the detection reagent (ATPLiteTM) with the FRD Workstation. The reaction was allowed to proceed at room temperature for 20 min and the signal was read in the luminescence mode on a ViewLux plate reader.
Table 1.
Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Reagent | 2 ml | 1 nM glCK in assay buffer |
| 2 | Reagent | 23 nl | Pin transfer compounds in DMSO |
| 3 | Time | 15 min | Room temperature incubation |
| 4a | Reagent | 1 ml | 600 mM CP, 600 mM ADP in assay buffer |
| 4b | Reagent | 1 ml | ATPLiteTM detection reagent |
| 5 | Time | 20 min | Room temperature incubation |
| 6 | Detection | ViewLux plate reader luminescence mode |
Counter-Screen Assay
The counter-screen assay was carried out in 1536-well format by replacing the glCK enzyme with ATP product at a concentration that was experimentally determined to be generated by the glCK assay conditions used. Briefly, 2 µl/well of 20 µM ATP in assay buffer was dispensed using the FRD Workstation. Compounds dissolved in DMSO were added from a source plate to the assay plate at a volume of 23 nl/well using an automated pin-tool station, and was allowed to equilibrate with the solution for 15 min at room temperature. Then, the detection reagent ATPLiteTM diluted 1:2 with assay buffer was dispensed as 2 µl/well with the FRD Workstation. The plates were incubated at room temperature for 5 min before the signal was read on a ViewLux plate reader.
Data Analysis
Z’ factor was calculated with the equation of Z’ = 1 – 3 (SDTotal + SDBasal) / (MeanTotal – MeanBasal), where SDTotal is the standard deviation of DMSO treated wells with 0.5 nM glCK, SDBasal is the standard deviation of no-enzyme control wells treated with DMSO, MeanTotal is the mean signal of DMSO treated wells with 0.1 nM glCK and MeanBasal is the mean signal of no-enzyme control wells treated with DMSO [20]. Coefficient of variation (CV) was calculated with the equation of CV = SDTotal / MeanBasal, expressed as a percentage. Data normalization and curve fitting was performed as previously described [21]. Briefly, raw plate reads for each titration point were first normalized relative to DMSO only wells (0% activity) and no enzyme control wells (100% activity), and then corrected by applying a pattern correction algorithm using compound-free control plates (DMSO plates). Concentration–response titration points for each compound were fitted to the Hill equation, yielding concentrations of half-maximal inhibition (IC50) and maximal response (efficacy) values. Data from the counter-screen underwent the same initial analysis, with the exception that raw luminescence counts were normalized relative to DMSO only wells (0% activity) and wells lacking ATP (100% activity).
Absorbance- Based Carbamate Kinase Assay
The coupled assay described previously [13] served as a confirmatory assay. The conversion of ADP + carbamoyl phosphate to ATP + ammonium carbamate was coupled to the conversion of NADP+ to NADPH using hexokinase, glucose-6-phosphate (G6P) dehydrogenase, and glucose. All chemical and biochemical components (except glCK) were purchased from Sigma-Aldrich. Briefly, the assay was performed in 96-well plates in 200 μL reaction solutions. 20 μL solutions containing 400 µM ADP, 400 µM carbamoyl phosphate, 30 mM MgCl2, 10 mM D-glucose, 1 mM NADP, 10 units hexokinase, 10 units G6P dehydrogenase, and 20 mM Tris-HCl (pH 7.5) were dispensed into the wells. Solutions containing glCK and 20 mM Tris-HCl (pH 7.5) were incubated for 15 minutes with different concentration of inhibitors, such that the final enzyme and inhibitor concentrations after adding 180µL aliquots to the wells were 5 nM glCK and 0.002, 0.02, 0.2, 1, 2, 20, and 200 μM inhibitor. The reactions were performed at room temperature and the reaction progress was monitored at 340 nm (Δε = 6.2 mM-1 cm-1) using a SPECTRAmax spectrophotometer (Molecular Devices Corp, Sunnyvale, California). IC50 values were calculated from the initial velocity data. Counter assays, omitting glCK and supplying ATP, were performed to confirm that the hexokinase and G6P dehydrogenase were not inhibited by the compounds.
RESULTS AND DISCUSSION
Assay Development and Optimization
In order to increase the assay sensitivity, a luminescence-based ATP content assay was applied to the forward CK reaction. The assay principles are illustrated in Fig. (1). The glCK enzyme converts the carbamoyl phosphate (CP) and ADP into ammonia, CO2 and ATP. The amount of ATP produced is then monitored via coupling to a firefly luciferase enzyme in the ATPLiteTM assay kit. Using this assay format, the kinetics of glCK were experimentally determined at room temperature for each of the substrates (Fig. 2a, b). The Km values of ADP and CP were 58.3 ± 2.4 µM and 52.5 ± 4.3 µM, respectively. These values are slightly lower than previously reported by an absorbance-based enzyme-coupled assay [13]. However, the previously reported values were determined at 37oC, while the current assay conditions room temperature. For the purpose of HTS, room temperature is preferred to prevent temperature gradients in assay plates associated with incubation at elevated temperatures. Therefore, to optimize the glCK assay for HTS, the steady-state kinetics was determined at room temperature.
Fig. (2).

Assay optimization. (a) Titration of ADP. The Km was determined with 2 mM CP and 2.5 nM glCK as 58.3 mM. (b) Titration of CP. The Km was determined with 333 mM ADP and 1.7 nM glCK as 52.5 mM. (c) The time course of the reaction was determined with 150 mM ADP, 150 mM CP and 0.5 nM glCK. The signal is linear up to 30 min incubation at room temperature.
In order for the assay to remain sensitive to competitive inhibitors while still providing wide signal range, the concentrations of substrates were kept at 150 µM each. A time course study of enzyme activity showed a linear increase in enzyme activity (0.5 nM glCK) up to 30 min at room temperature (Fig. 2c). An incubation time of 20 min was then selected as optimal condition that falls within the linear region of the time course curve and yields sufficient signal.
Assay Miniaturization and Validation
The glCK assay was miniaturized to 1536-well plate format from the initial 384-well low volume plate by a 2-fold reduction in assay volume. Because DMSO is the solvent of all compounds screened, a DMSO plate was used initially to validate the 1536-well assay. The signal-to-basal ratio (S/B), Z’ factor and correlation of variation (CV) obtained from this DMSO plate test were 11.5-fold, 0.8 and 5.7%, respectively (Fig. 3). These values indicate that the assay in 1536-well plate format is robust for HTS.
Fig. (3).

Assay miniaturization. (a) Plate map for 1536-well format. Columns 1-2: no enzyme DMSO control, columns 3-4: 0.5 nM glCK DMSO control and columns 5-48: 0.5 nM glCK compound area. (b) Scatter plot of DMSO plate test. DMSO was pin-transferred as 23 nl/well as a solvent control.
Primary Screen and Hit Follow-Up
Using these optimized conditions, the glCK assay was used to screen both the LOPAC1280 and NPC libraries in quantitative HTS (qHTS) format [21]. The screen was carried out at a 5-concentration titration ranging from 92 nM to 57.5 µM. To eliminate false positive compounds that affect the assay detection reagents, a counter-screen assay that consisted of the glCK reaction product ATP and the ATPLite detection reagent without the glCK enzyme was screened in parallel. From assay calibration against known ATP concentration titrations, we found that the glCK assay generated 20 µM ATP, which was applied to the counter-screen. The parallel screens of both the primary and counter-screen assays revealed concentration-response curves for all the primary hits, and provided a rich source of information for potency, efficacy, and false positive hits. Using the hit selection criteria of having IC50 < 10 µM and > 70% maximal inhibition in the glCK assay with <30% maximal inhibition in the counter-screen assay, a total of 37 primary hits were selected with a hit rate was 0.9 %.
These 37 hits were cherry picked and retested in the glCK and counter-assay in a 1:3 titration for 12 concentrations for the measurements of IC50 values. A total of 30 compounds out of 37 primary hits were confirmed (Table 2). Of the confirmed hits, three compounds showed sub-micromolar potencies (Fig. 4). It is interesting to note that several structural scaffolds were repeatedly found to be active. A class of chelator compounds that included disulfiram, thiram and sodium diethyldithiocarbamate inhibited glCK with IC50 values of 0.5 µM, 4.1 µM and 16.3 µM respectively (Table 2). In addition a second class of molecules containing a polyphenol scaffold was also found among the hits (Table 2). Polyphenols belong to a well-known class of general kinase inhibitors, but their activity on carbamate kinase has not been reported elsewhere. Many of the polyphenolic inhibitors identified in this screen are dopamine mimetics, and have previously reported activity as dopamine receptors modulators. Since Giardia infection is limited to the intestine, this potential off-target activity could theoretically be eliminated by synthesizing analogs with less systematic absorption to increase local drug concentration in gastrointestinal tract.
Table 2.
Confirmed Hits in the Luminescence Assay
| Structure | Compound Name (NCGC ID) | IC50 (mM) | Efficacy (% Inhibition) |
|---|---|---|---|
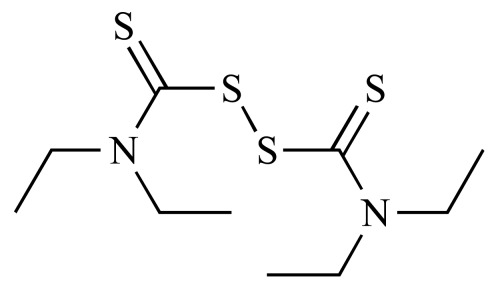 |
Disulfiram (NCGC00016000-03) | 0.58 | 90.5 |
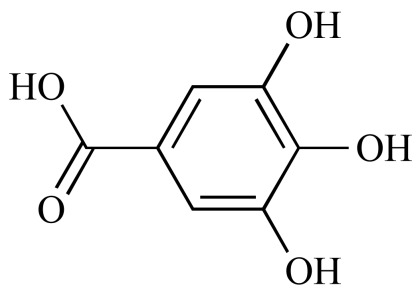 |
Gallic acid (NCGC00091125-01) | 0.65 | 89.2 |
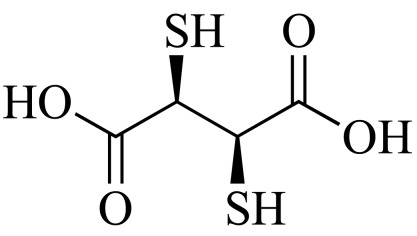 |
Succimer (NCGC00013217-01) | 0.65 | 85.5 |
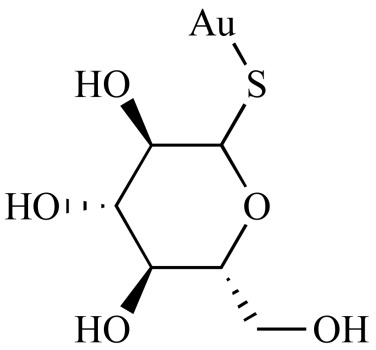 |
Aurothioglucose (NCGC00096015-01) | 1.46 | 81.2 |
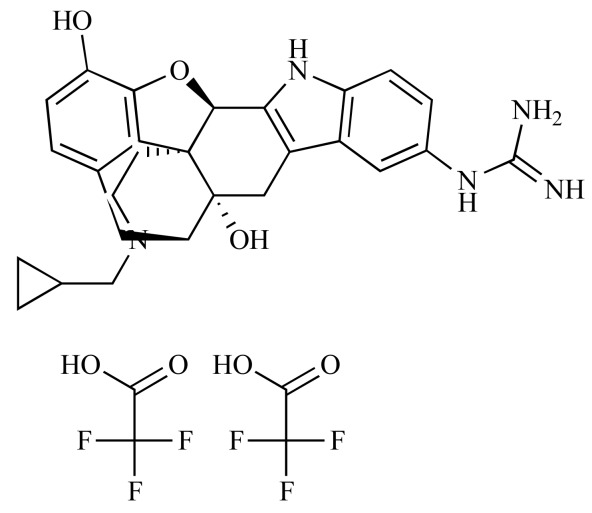 |
GNTI di-trifluoroacetate (NCGC00093944-01) | 1.83 | 86.0 |
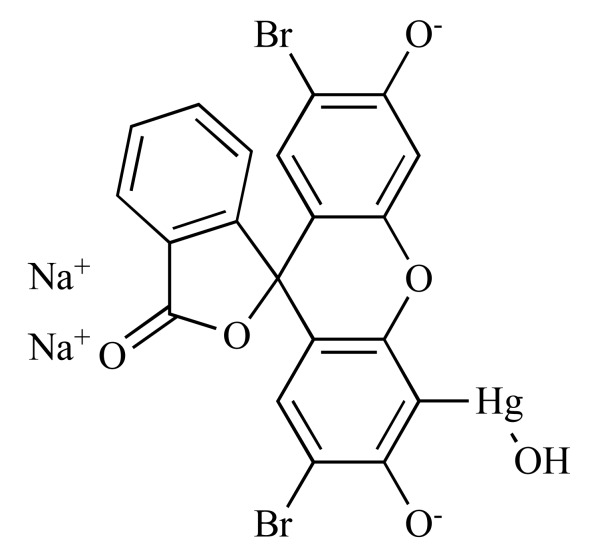 |
Mercurochrome (NCGC00017018-01) | 2.06 | 93.8 |
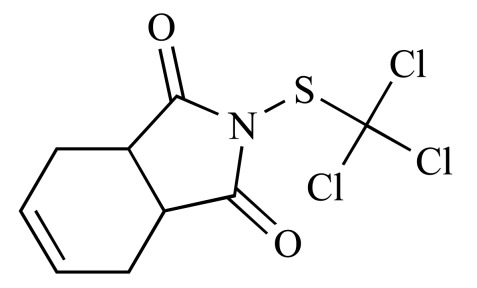 |
Captan (NCGC00091034-02) | 2.59 | 88.6 |
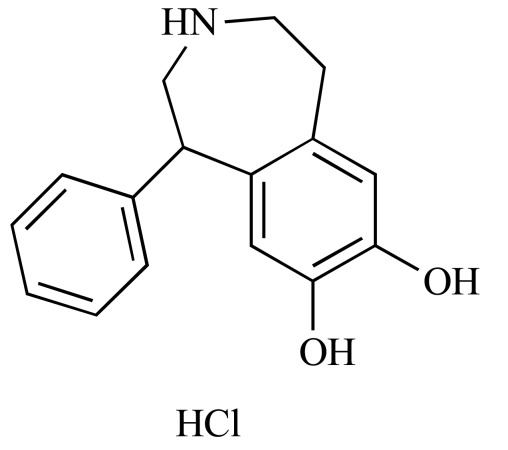 |
(+/-)-SKF-38393 hydrochloride (NCGC00093856-01) | 3.26 | 91.2 |
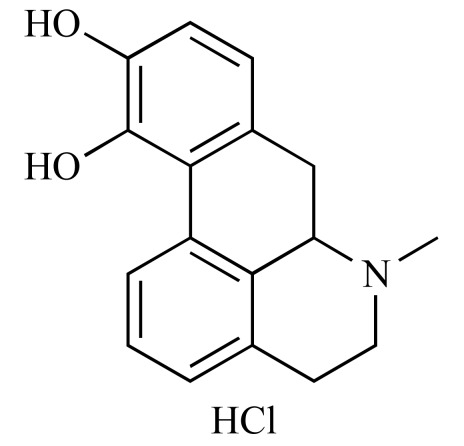 |
Apomorphine hydrochloride hemihydrate (NCGC00093584-01) | 3.26 | 89.4 |
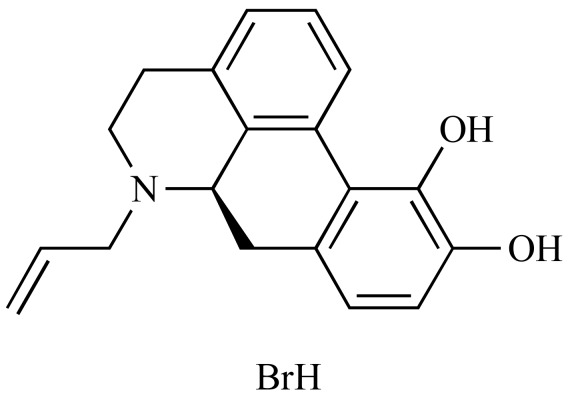 |
R(-)-N-Allylnorapomorphine hydrobromide (NCGC00093832-01) | 3.66 | 89.0 |
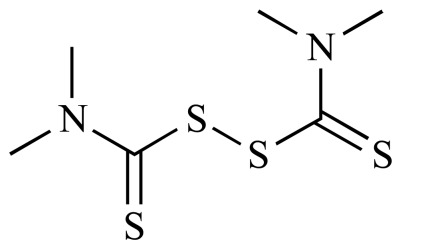 |
Thiram (NCGC00091563-01) | 4.11 | 92.3 |
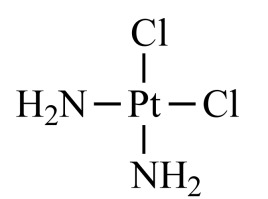 |
Cisplatin (NCGC00090759-01) | 5.17 | 88.2 |
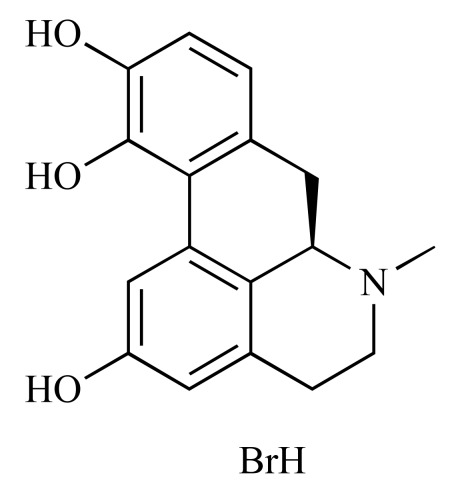 |
R(-)-2,10,11-Trihydroxyaporphine hybrobromide (NCGC00093861-01) | 5.17 | 90.7 |
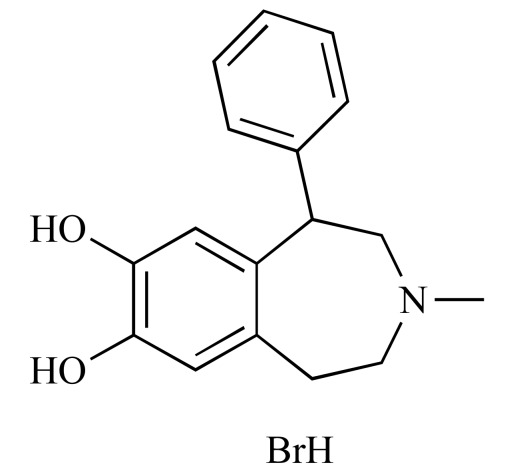 |
SKF 75670 hydrobromide (NCGC00094362-01) | 5.80 | 85.4 |
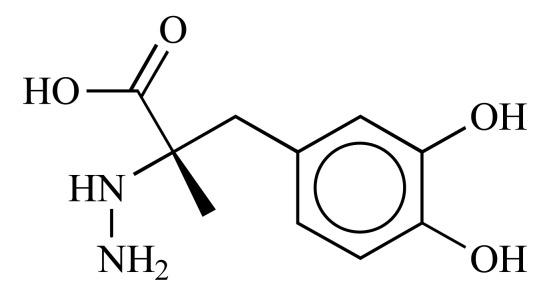 |
Carbidopa (NCGC00024596-05) | 5.80 | 79.4 |
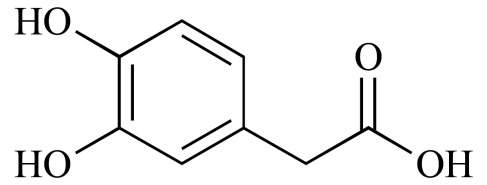 |
3,4-Dihydroxyphenylacetic acid (NCGC00015381-02) | 6.51 | 80.2 |
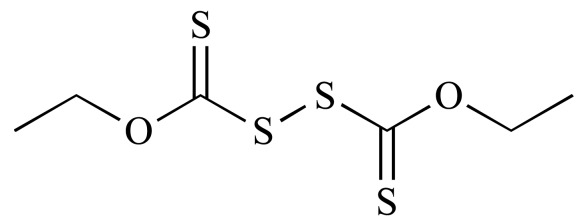 |
Bis(ethylxanthogen) (NCGC00160587-01) | 6.51 | 82.3 |
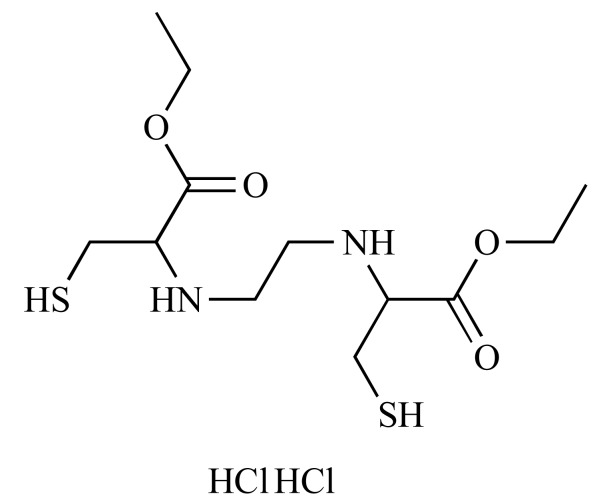 |
N,N'-1,2-Ethylenediylbiscysteine, diethyl ester dihydrochloride (NCGC00167479-01) | 8.19 | 79.4 |
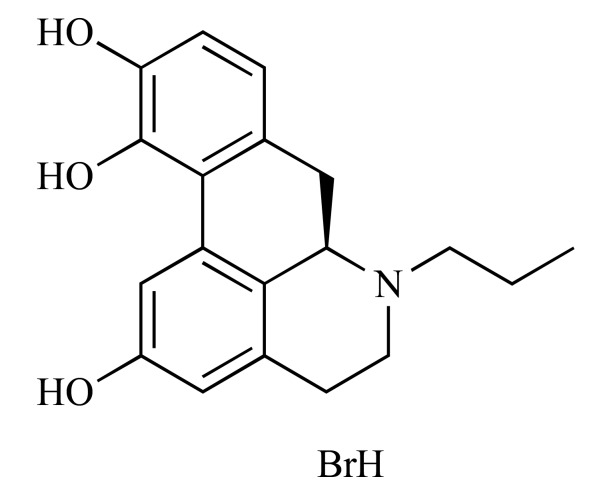 |
R(-)-2,10,11-Trihydroxy-N-propylnoraporphine hydrobromide (NCGC00093870-01) | 8.19 | 86.8 |
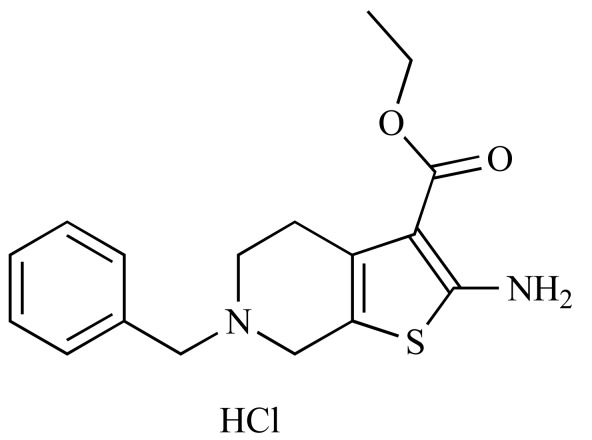 |
Tinoridine hydrochloride (NCGC00159451-02) | 10.31 | 80.2 |
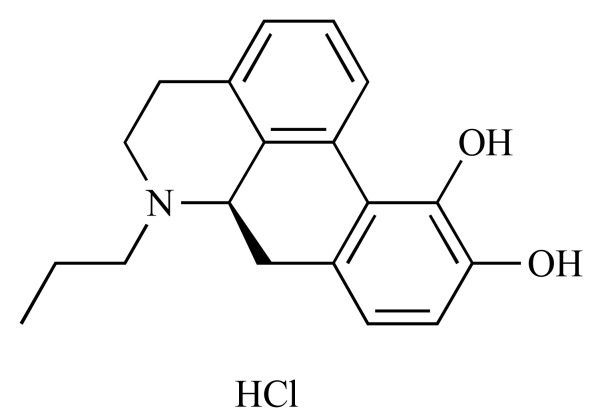 |
R(-)-Propylnorapomorphine hydrochloride (NCGC00093855-01) | 10.31 | 89.9 |
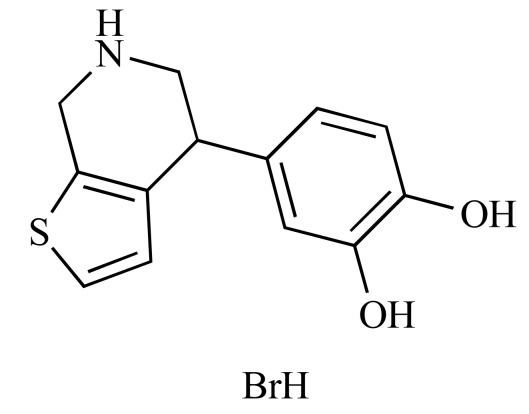 |
SKF 89626 (NCGC00094378-01) | 10.31 | 87.4 |
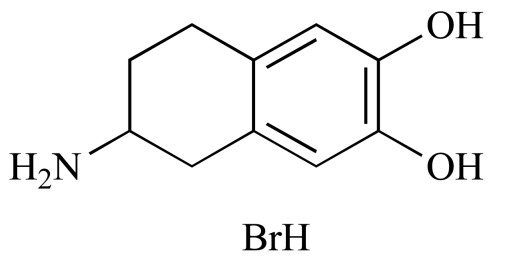 |
6,7-ADTN hydrobromide (NCGC00093839-01) | 12.98 | 83.2 |
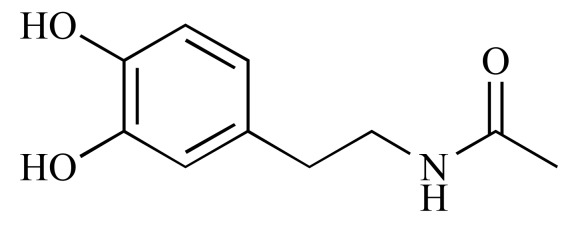 |
N-Acetyldopamine monohydrate (NCGC00015101-02) | 12.98 | 77.3 |
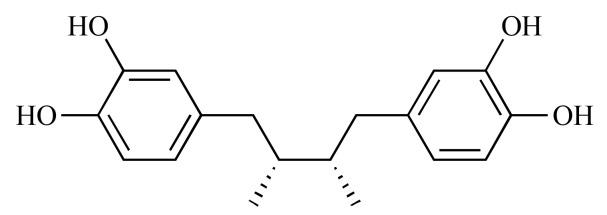 |
Nordihydroguaiaretic acid (NCGC00094201-01) | 12.98 | 81.3 |
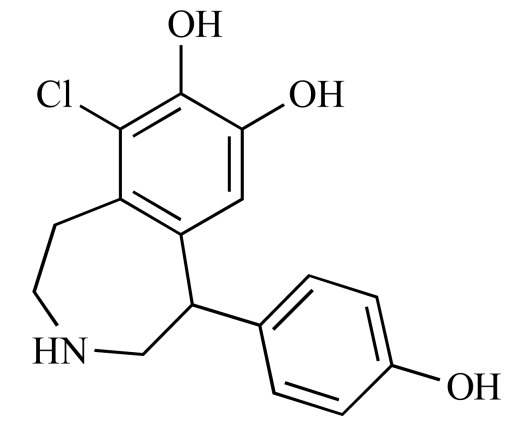 |
Fenoldopam (NCGC00015444-04) | 16.34 | 84.5 |
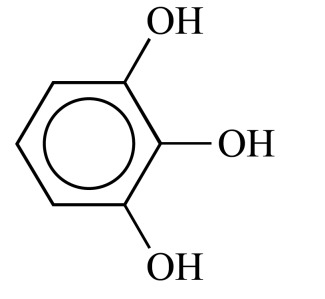 |
Pyrogallol (NCGC00091507-01) | 16.34 | 77.4 |
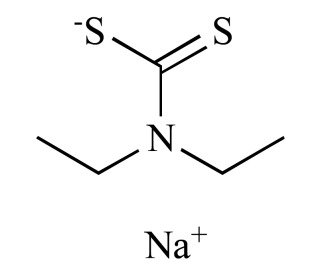 |
Ditiocarb (NCGC00166328-01) | 16.34 | 77.7 |
 |
Adefovir dipivoxil (NCGC00164624-01) | 16.34 | 83.6 |
 |
Dihydrexidine hydrochloride (NCGC00093812-01) | 20.58 | 82.9 |
Fig. (4).

Concentration response curves for (a) Disulfiram, with IC50 = 0.58 mM, (b) Gallic acid, with IC50 = 0.65 mM and (c) Succimer, with IC50 = 0.65 mM in the glCK assay.
Secondary Assay Follow-Up
Of the 30 hits confirmed by the luminescence assay, 13 compounds were readily available for purchase and were tested with a secondary assay employing a different coupling enzymes and readout (UV spectroscopy). Of these, 4 compounds demonstrated good correlation between the two assays, with less than 2-fold difference in IC50 values, 5 compounds showed less than 10-fold difference in IC50 values, and one compound had a 17-fold difference between the two IC50 values (Table 3). The latter three compounds were not confirmed in the absorbance assay, with IC50 values evaluated as greater than 200 μM, the maximum concentration used in the assays (Table 3). The differences in inhibitor potency might be attributed in part to significant differences between the assays. For example, the substrate concentration was 150 µM in the luminescence assay versus 40 µM in the absorbance assay, and enzyme was concentration 0.5 nM in the luminescence assay versus 5 nM in the absorbance assay. The higher enzyme concentration in the secondary assay was necessary to detect the absorbance signal. Considering these differences, 9 out of 13 compounds showed IC50 values with less than 10-fold difference between the two assays. Two of the top compounds, disulfiram and succimer showed good confirmation in potencies and have submicromolar IC50 values in both assays (Table 3).
Table 3.
Secondary Assay Results
| Compound Name (NCGC ID) | IC50 Luminescence Assay (mM) | IC50 Absorbance Assay (mM) |
|---|---|---|
| Disulfiram (NCGC00016000-03) | 0.58 | 1.39 |
| Thiram (NCGC00091563-01) | 4.11 | 0.64 |
| Mercurochrom (NCGC00017018-01) | 2.06 | 0.22 |
| Succimer (NCGC00013217-01) | 0.65 | 0.54 |
| SKF-38393 hyrochloride (NCGC00093856-01) | 3.26 | 1.79 |
| Captan (NCGC00091034-02) | 2.59 | 0.47 |
| Gallic acid (NCGC00091125-01) | 0.65 | 11.42 |
| Pyrogallol (NCGC00091507-01) | 16.34 | 2.70 |
| 3,4-Dihydroxyphenylacetic acid (NCGC00015381-02) | 6.51 | 11.73 |
| 6,7-ADTN hydrobromide (NCGC00093839-01) | 12.98 | 15.69 |
| Adefovir dipivoxil (NCGC00164624-01) | 16.34 | >200 |
| Carbidopa (NCGC00024596-05) | 5.80 | >200 |
| Apomorphine hydrochloride hemihydrate (NCGC00093584-01) | 3.26 | >200 |
CONCLUSIONS
While several CK assays have been reported, they are not applicable for HTS due to limitations in assay sensitivity and throughput. Previous studies have used these assays to characterize CK enzymes purified from various microorganisms, but did not screen for enzyme inhibitors in a systematic fashion. We have described here the development of a new luminescence-based assay for the forward CK reaction that was successfully validated in a pilot screen of 4096 small molecule compounds in 1536-well format, resulting in a total of 30 confirmed glCK inhibitors.
A Giardia viability assay was developed previously and had been used to screen the same compound libraries [22]. Of the confirmed compounds found in this glCK screen, only disulfiram, thiram, and ditiocarb were also active in the previous Giardia killing screen. The other compounds failed to show activity (>30% reduction in ATP content) in killing Giardia at the maximum screening concentration of 38.3 µM. Furthermore, thiram and disulfiram have been previously shown to have anti-giardial activities in a mouse model for Giardia infection [23]. It is possible that the other 28 hits were inactive against the parasite because they were not able to cross the cell wall and/or membrane of Giardia. It is also possible that some were metabolized by the organism to form an inactive product. Currently, there is much discussion regarding the merits of phenotypic versus target based screening methods. While the phenotypic Giardia killing assay directly identifies physiologically active compounds, the targets of these compounds are unclear, making it difficult to improve compound potencies through medicinal chemistry. Therefore, the luminescence CK assay described here offers an alternative drug development strategy that is target-based. The chemical optimization of other structural classes of these confirmed glCK inhibitors such as the polyphenoic compounds may improve the permeability and stability in Giardia trophozoites as required for treatment of giardiasis. Moreover, to the best of our knowledge, these are the first CK inhibitors reported in the literature other than a non-hydrolyzable ATP, which is a very weak mM-range inhibitor [13]. The glCK inhibitors reported here could be useful as research tools. In addition, since the arginine dihydrolase pathway has been found to be an important metabolic pathway in multiple microorganisms, the CK luminescence assay described here could serve as a common platform to assay CK enzymes of other species.
ACKNOWLEDGEMENT
This study was supported by National Institutes of Health grant R56 AI059733 (to O.H.), the Molecular Libraries Initiative of the NIH Roadmap for Medical Research, and the Intramural Research Program of National Center for Advancing Translational Sciences, National Institutes of Health.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
- 1.Upcroft JA, Upcroft P. Drug susceptibility testing of anaerobic protozoa. Antimicrob Agents Chemother. 2001;45(6 ):1810–4. doi: 10.1128/AAC.45.6.1810-1814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright JM, Dunn LA, Upcroft P, et al. Efficacy of antigiardial drugs. Expert Opin Drug Saf. 2003;2(6 ):529–41. doi: 10.1517/14740338.2.6.529. [DOI] [PubMed] [Google Scholar]
- 3.McPhee SJ, Papadakis MAE. Current medical diagnosis & treatment 2010. The McGraw-Hill Companies, Inc; 2010. [Google Scholar]
- 4.Farbey MD, Reynoldson JA, Thompson RC. In vitro drug susceptibility of 29 isolates of Giardia duodenalis from humans as assessed by an adhesion assay. Int J Parasitol. 1995;25(5 ):593–9. doi: 10.1016/0020-7519(94)00174-m. [DOI] [PubMed] [Google Scholar]
- 5.Upcroft JA, Upcroft P, Boreham PF. Drug resistance in Giardia intestinalis. Int J Parasitol. 1990;20(4 ):489–96. doi: 10.1016/0020-7519(90)90196-t. [DOI] [PubMed] [Google Scholar]
- 6.Crow VL, Thomas TD. Arginine metabolism in lactic streptococci. J Bacteriol. 1982;150(3 ):1024–32. doi: 10.1128/jb.150.3.1024-1032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Pritchard GG, Hardman MJ, et al. Occurrence of arginine deiminase pathway enzymes in arginine catabolism by wine lactic acid bacteria. Appl Environ Microbiol. 1995;61(1 ):310–6. doi: 10.1128/aem.61.1.310-316.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yarlett N, Martinez MP, Moharrami MA, et al. The contribution of the arginine dihydrolase pathway to energy metabolism by Trichomonas vaginalis. Mol Biochem Parasitol. 1996;78(1-2 ):17–25. doi: 10.1016/s0166-6851(96)02616-3. [DOI] [PubMed] [Google Scholar]
- 9.Edwards MR, Knodler LA, Wilson JR, Schofield PJ. Arginine metabolism during culture of Giardia intestinalis. Mol Biochem Parasitol. 1992;53(1-2 ):97–103. doi: 10.1016/0166-6851(92)90011-8. [DOI] [PubMed] [Google Scholar]
- 10.Schofield PJ, Edwards MR, Mathews J, Wilson JR. The pathway of arginine catabolism in Giardia intestinalis. Mol Biochem Parasitol. 1992;51(1 ):29–36. doi: 10.1016/0166-6851(92)90197-r. [DOI] [PubMed] [Google Scholar]
- 11.Palm JE, Weiland MEL, Griffiths WJ, Ljungström I, Svärd SG. Identification of immunoreactive proteins during acute human giardiasis. J Infect Dis. 2003;187(12 ):1849–59. doi: 10.1086/375356. [DOI] [PubMed] [Google Scholar]
- 12.Vincendeau P, Gobert AP, Daulouède S, Moynet D, Mossalayi MD. Arginases in parasitic diseases. Trends Parasitol. 2003;19(1 ):9–12. doi: 10.1016/s1471-4922(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 13.Galkin A, Kulakova L, Wu R, Nash TE, Dunaway-Mariano D, Herzberg O. X-ray structure and characterization of carbamate kinase from the human parasite Giardia lamblia. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(4 ):386–90. doi: 10.1107/S1744309110004665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalman SM, Duffield PH. Purification And Properties Of Carbamate Kinase From Streptococcus Faecalis. Biochim Biophys Acta. 1964;92:498–512. doi: 10.1016/0926-6569(64)90010-0. [DOI] [PubMed] [Google Scholar]
- 15.Marina A, Uriarte M, Barcelona B, et al. Carbamate kinase from Enterococcus faecalis and Enterococcus faecium--cloning of the genes, studies on the enzyme expressed in Escherichia coli, and sequence similarity with N-acetyl-L-glutamate kinase. Eur J Biochem. 1998;253(1 ):280–91. doi: 10.1046/j.1432-1327.1998.2530280.x. [DOI] [PubMed] [Google Scholar]
- 16.Lea WA, Xi J, Jadhav A, et al. A high-throughput approach for identification of novel general anesthetics. PLoS One. 2009;4(9 ):e7150. doi: 10.1371/journal.pone.0007150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Titus SA, Li X, Southall N, et al. A cell-based PDE4 assay in 1536-well plate format for high-throughput screening. J Biomol Screen. 2008;13(7 ):609–18. doi: 10.1177/1087057108319977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang R, Southall N, Wanget Y, et al. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3(80 ):80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yasgar A, Shinn P, Jadhav A, et al. Compound Management for Quantitative High-Throughput Screening. JALA Charlottesv Va. 2008;13(2 ):79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2 ):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 21.Inglese J, Auld DS, Jadhav A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA. 2006;103(31 ):11473–8. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CZ, Kulakova L, Southall N, et al. High-throughput giardia lamblia viability assay using bioluminescent atp content measurements. Antimicrob Agents Chemother. 2011;b(2 ):667–75. doi: 10.1128/AAC.00618-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nash T, Rice WG. Efficacies of zinc-finger-active drugs against Giardia lamblia. Antimicrob Agents Chemother. 1998;42(6 ):1488–92. doi: 10.1128/aac.42.6.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]