

Abstract
Intestinal ischemia-reperfusion (IR)-induced injury results from a complex cascade of inflammatory components. In the mouse model of intestinal IR, the serum protein, β2-glycoprotein I (β2-GPI) binds to the cell surface early in the cascade. The bound β2-GPI undergoes a conformational change which exposes a neoantigen recognized by naturally occurring antibodies and initiates the complement cascade. We hypothesized that providing additional antigen with exogenous β2-GPI would alter IR-induced tissue injury. Administration of human but not mouse β2-GPI attenuated IR-induced tissue damage and prostaglandin E2 production indicating a physiological difference between β2-GPI isolated from the two species. To investigate whether structural features were responsible for this physiological difference, we compared the chemical, physical and biochemical properties of the two proteins. Despite possessing 76% amino acid identity and 86% sequence homology, we found that mouse β2-GPI differs from the human protein in size, carbohydrate chain location, heterogeneity and secondary structural content. These data suggest that the structural differences result in mouse Ab recognition of soluble human but not mouse β2-GPI and attenuated IR-induced injury. We conclude that caution should be exercised in interpreting results obtained by using human β2-GPI in a mouse model.
Keywords: Apolipoprotein H, Prostaglandin E2, Mouse, Ischemia
1. Introduction
Ischemia induces tissue damage which is exacerbated during subsequent tissue reperfusion. Multiple studies identified a sequence of events that lead to increased injury and inflammation. The sequence requires formation of a cell bound antigen/antibody complex resulting in excessive complement activation and prostaglandin E2 (PGE2) production (Austen et al., 1999; Fruchterman et al., 1998; Hill et al., 1992; Moses et al., 2009). Previous studies indicated that complement activation is required for intestinal injury with all three complement initiating pathways playing a role (Chen et al., 2009; Hart et al., 2005; Williams et al., 1999). Other studies indicated that PGE2 production is necessary but not sufficient for ischemia/reperfusion (IR)-induced intestinal damage (Moses et al., 2009; Pope et al., 2010). The requirement for naturally occurring antibodies (Ab) in IR-induced, complement-mediated tissue injury and PGE2 production was previously established (Weiser et al., 1996; Williams et al., 1999; Sparkes et al., 2010). Together these studies present a complex pathogenic cascade of inflammatory components triggering IR-induced tissue damage.
In contrast to the mechanisms of injury, the specific antigens recognized by the Ab are not as clearly defined. Naturally occurring Ab frequently bind with low affinity and cross react with multiple antigens (Avrameas, 1991; Martin and Kearney, 2000). Multiple antigen/antibody complexes have been identified as initiators of IR-induced injury, including non-muscle myosin and annexin IV (Kulik et al., 2009; Zhang et al., 2004). Previously, we demonstrated that naturally occurring anti-β2 glycoprotein I (β2-GPI) Ab are instrumental in intestinal IR-induced tissue damage (Fleming et al., 2004). In addition, peptides derived from the mouse β2-GPI sequence attenuated β2-GPI binding to cell membranes and subsequent intestinal injury in the mouse model of intestinal IR (Fleming et al., 2010). Together, these data suggest that natural Ab recognition of cell surface bound β2-GPI is critical to IR-induced tissue damage.
β2-GPI, also referred to as apolipoprotein H, is a heavily glycosylated, 43 kDa protein found in the plasma (Gries et al., 1989). Purified human β2-GPI contains multiple glycosylation sites with varied sialic acid content yielding 5–7 species with different isoelectric points (Gries et al., 1989). The 3D structure of membrane-bound human β2-GPI indicates five domains in a J-shaped chain (open conformation) whereas the soluble protein appears to be a condensed ring-like structure (closed conformation) (Agar et al., 2010; Hammel et al., 2002; Schwarzenbacher et al., 1999). Importantly, Ab recognition of the protein requires neoantigen exposure that occurs when β2-GPI binds to a cell surface and takes on the J-shaped chain conformation (Fig. 1A). Domain 5 contains three disulfide bonds, a lysine rich peptide sequence and a hydrophobic quadruplet (Bouma et al., 1999) which interact and bind with amphipathic anionic molecules such as phosphatidylserine exposed on early apoptotic cells (Balasubramanian et al., 1997, 2005).
Fig. 1.

Hypothetical models of exogenous β2-GPI-Ab interactions. Under normal conditions, the closed conformation of soluble β2-GPI is not recognized by anti-β2-GPI Ab (A). During IR-induced stress, membrane lipid alterations allow Ab recognition of bound β2-GPI (mouse protein is dark, human protein is light) and induces injury which may be exacerbated by exogenous β2-GPI (B). Excess β2-GPI may bind to altered lipids without Ab recognition (C) or exogenous β2-GPI may form soluble immunocomplexes resulting in attenuated IR-induced injury (D).
Despite the relatively high concentration of β2-GPI in the plasma (4–5 μM) (Agar et al., 2011b), specific physiological roles for the protein are not yet well defined. However, human β2-GPI is clearly involved in the autoimmune diseases, anti-phospholipid syndrome and systemic lupus erythematosus (Cabiedes et al., 1995). Multiple activities have been ascribed to β2-GPI, such as binding to cellular debris with exposed negative charges, nucleic acids, apoptotic bodies and endothelial microvesicles (Hagihara et al., 2002; Manfredi et al., 1998). Recent studies demonstrated that β2-GPI scavenges lipopolysaccharide from the blood stream (Agar et al., 2011a,b; Fischetti et al., 2005; Laplante et al., 2011; Meroni et al., 2001; Tolomeo et al., 2009); moreover, β2-GPI also binds to Streptococcal protein H (Nilsson et al., 2008) and other pathogenic derived proteins (Stefas et al., 2011).
As β2-GPI-derived peptides attenuate deposition of both β2-GPI and anti-β2-GPI Ab in response to IR, it is possible that administration of exogenous β2-GPI may alter IR-induced injury. As indicated in Fig. 1, additional β2-GPI may be deposited on the cell surface leading to increased Ab binding, complement activation and subsequent injury (Fig. 1B). It is also possible that purification of the proteins structurally alters β2-GPI preventing Ab recognition of cell surface bound β2-GPI and resulting in attenuated injury (Fig. 1C). Finally, purification of β2-GPI may change the conformation and allow formation of soluble immunocomplexes (Fig. 1D) which precludes Ab recognition of endogenous β2-GPI and results in attenuated IR-induced tissue injury.
We initially tested the overall hypothesis that treating with β2-GPI would alter intestinal injury by injecting mice with purified β2-GPI prior to inducing Sham or IR-induced injury. Neither human β2-GPI nor the mouse protein exacerbated tissue damage. Surprisingly, human, but not mouse β2-GPI protein, attenuated IR-induced tissue damage and PGE2 production, suggesting that a structural and/or physiological difference of soluble human β2-GPI prevents IR-induced injury in the mouse. Despite possessing 76% amino acid identity and 86% sequence homology, we further demonstrate that human and mouse β2-GPI contain distinct chemical, physical and biochemical properties which contribute to the differences observed in IR-induced injury and inflammation in the mouse.
2. Methods
2.1. Mice
C57Bl/6, Balb/c, and Rag-1−/− mice were obtained from Jackson Laboratories and bred in the Division of Biology at Kansas State University with free access to food and water. Mice were maintained in a specific pathogen free environment (Helicobacter species, mouse hepatitis virus, minute virus of mice, mouse parvovirus, Sendai virus, murine norovirus, Mycoplasma pulmonis, Theiler’s murine encephalomyelitis virus, and endo- and ecto-parasites). Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and was approved by the Institutional Animal Care and Use Committee at Kansas State University.
2.2. Ischemia/reperfusion
Animals were subjected to IR similar to previously described studies (Moses et al., 2009). Briefly, ketamine (16 mg/kg) and xylazine (80 mg/kg) anesthetized male mice (2–4 months old) were administered buprenorphine (0.06 mg/kg) for pain. After laparotomy and a 30 min equilibration period, ischemia was induced by application of a small vascular clamp (Roboz Surgical Instruments, Gaithersburg, MD) to the isolated superior mesenteric artery. Ischemia was visually confirmed by blanching of the mid-jejunum. The bowel was covered with moistened surgical gauze to prevent desiccation. After 30 min of ischemia, the clamp was removed and the intestines reperfused for 2 h. Reperfusion was confirmed by observing a pinkish color change of the bowel and the return of pulsatile flow to the mesenteric artery and its branches. Some mice received β2-GPI (either purified or purchased from Aro Tec (Wellington, New Zealand) or Fitzgerald (Acton, MA)) i.v. 30 min prior to ischemia. Sham-treated animals underwent the same surgical intervention without occlusion. All procedures were performed with the animals breathing spontaneously and body temperature maintained at 37 °C using a water-circulating heating pad. Additional ketamine and xylazine was administered as needed and immediately prior to sacrifice. After sacrifice, 2 cm sections of the small intestine, 10 cm distal to the gastroduodenal junction were harvested for histologic evaluation and PGE2 determination.
2.3. Histology and immunohistochemistry
Small intestine specimens were promptly fixed in 10% buffered formalin phosphate and embedded in paraffin, sectioned transversely (8 μm), and H&E stained. The mucosal injury score (SMI) was graded on a six-tiered scale modified from Chiu et al. (1970). Briefly, the average damage score of the intestinal section (75–150 villi) was determined after grading each villus from 0 to 6. Normal villi were assigned a score of zero; villi with tip distortion were assigned a score of 1; a score of 2 was assigned when Guggenheims’ spaces were present; villi with patchy disruption of the epithelial cells were assigned a score of 3; a score of 4 was assigned to villi with exposed but intact lamina propria with epithelial sloughing; a score of 5 was assigned when the lamina propria was exuding; last, villi that displayed hemorrhage or were denuded were assigned a score of 6. Photomicrographs were obtained from H&E stained slides using a 20×, 0.5 Plan Fluor objective on Nikon 80i microscope and images acquired at room temperature using a Nikon DS-5 M camera with DS-L2 software. Additional intestinal sections were snap frozen in OCT freezing medium and 8 μm cryosections cut for analysis of C3 deposition as described previously (Fleming et al., 2010). Briefly, acetone fixed slides were blocked with 10% normal donkey sera for 30 min at 37 °C prior to staining with rat anti-mouse C3 Ab (Hycult Biotechnologies, Plymouth Meeting, PA) for 1 hr at room temperature and followed by a Texas-red conjugated donkey-anti-rat IgG secondary Ab (Jackson Immunoresearch, West Grove, PA). Each experiment contained serial sections stained with the appropriate isotype control Ab. All slides were mounted with ProLong Gold (Invitrogen, Grand Island, NY). A blinded observer obtained images at room temperature using a Nikon eclipse 80i microscope equipped with a CoolSnap CF camera (Photometrics, Tucson, AZ) and analyzed using Metavue software (Molecular Devices, Sunnyvale, CA).
2.4. Eicosanoid and cytokine determination
The ex vivo generation of eicosanoids from small intestinal tissue was determined as described previously (Sjogren et al., 1994). Briefly, fresh mid-jejunum sections were minced, washed and resuspended in 37 °C oxygenated Tyrode’s buffer (Sigma, St. Louis, MO). After incubating for 20 min at 37 °C, supernatants were collected and supernatants and tissue were stored at −80 °C until assayed. The concentration of prostaglandin E2 (PGE2) was determined using an enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI). The tissue protein content was determined using the bicinchoninic acid assay (Pierce, Rockford, IL) adapted for use with microtiter plates. PGE2 production was expressed per mg protein per 20 min.
2.5. β2-GPI purification
Human and mouse (C57Bl/6, Balb/c or Rag-1−/−) β2-GPI were purified using the procedure described and modified previously (Schultze, 1961; Wurm, 1984). Briefly, perchloric acid was added dropwise to ice cold mouse or human citrated plasma to a final concentration of 1.75% prior to centrifugation at 4000 rpm at 4 °C. The supernatant was then neutralized with saturated sodium carbonate and 1.0 M acetic acid prior to dialyzing overnight against 30 mM NaCl 20 mM Tris, pH 8.0, at 4 °C with at least 2 buffer changes. The dialyzed solution was applied to a HiTrap Heparin HP column (ThermoFisher, USA), washed with dialysis buffer and eluted with 175–200 mM NaCl 20 mM Tris, pH 8.0.
2.6. Molecular weight analysis of β2-GPI
MALDI-TOF MS was performed on a Bruker Ultraflex II TOF/TOF mass spectrometer.
Molecular weight of purified β2-GPI was analyzed in the positive mode. Sinapinic acid was used as a matrix. MS spectra were recorded in linear mode within a mass range from m/z 10,000 to m/z 70,000 and externally calibrated using a protein mass standard kit II (Bruker Daltonics, Billerica, MA).
2.7. Proteomics analysis using in gel digestion
Coomassie brilliant blue stained and excised gel pieces were destained by several 10 min incubations in 100 μL of 50% acetonitrile (ACN) at 30 °C. After destaining, reduction and alkylation of cysteine residues was accomplished with Tris[2-carboxyethyl]phosphine (TCEP) and Iodoacetamide followed by the supplier protocol (#8971 in gel trypsin digestion kit, Thermo Scientific, USA). After washing the gels with 50% ACN, 100% ACN (50 μL) was added for 10 min at 30 °C to shrink the gel pieces. After discarding the solvent and drying by speed vacuum concentration, the gel plugs were rehydrated in 20 μL of sequencing grade trypsin (200 ng; Trypsin Gold, Promega, Madison, WI) in 20 mM ammonium bicarbonate. Upon rehydration, the gel plugs were incubated in an additional 20 μL of 20 mM ammonium bicarbonate with 10% ACN at 30 °C for 17 h. Tryptic peptides were extracted from the gel plugs with 100 μL of 50% ACN containing 2% trifluoroacetic acid (TFA) at 30 °C for 30 min. Extracted peptides were concentrated by speed vacuum concentration.
2.8. Nano-HPLC and electrospray ionization tandem mass spectrometry (ESI–MS/MS)
Nano-HPLC was performed automatically using a microcolumn switching device (Switchos; LC Packings) coupled to an autosampler (Famos: LC Packings) and a nanogradient generator (UltiMate Nano HPLC; LC Packings). Peptide solution (30 μL) was loaded on a C18 reversed-phase capillary column (75 μm ID × 15 cm, PepMap: Dionex) in conjunction with an Acclaim C18 PepMap trapping column (300 μm id × 10 mm, Dionex). Peptides were separated by a nanoflow linear ACN gradient using buffer A (0.1% formic acid, 2% ACN) and buffer B (0.1% formic acid, 80% ACN) starting from 5% buffer B to 70% over 55 min at a flow rate of 200 nL/min. Then column was washed by 95% of buffer B for 5 min. The system control software, Hystar 3.2, was used to control the entire process. The eluted peptides were injected into an HCT Ultra Ion Trap Mass Spectrometer (Bruker Daltronics). The mass spectrometer was set up in the data dependent MS/MS mode to alternatively acquire full scans (m/z acquisition range from 300 to 1700 Da). The four most intense peaks in any full scan were selected as precursor ions and fragmented by collision energy. MS/MS spectra were interpreted and peak lists were generated by DataAnalysis 3.4 and Biotools 3.0 software (Bruker Daltronics).
2.9. Bioinformatics
Peptide masses were compared to Swiss Prot Database using MASCOT 2.2 (http://www.matrixscience.com). Human or mouse genomes were selected for the taxonomy search. The following parameters were used in all searches: the maximum number of missed cleavages allowed was 2; the mass tolerance was 0.6 Da for MS and 0.6 Da for MS/MS. Fixed modification was set on cysteine with carbamidomethylation. Variable modification was based on methionine with oxidation. Positive protein identifications using a threshold of 0.05 were used. Peptides scoring <20 were automatically rejected, ensuring all protein identifications were based on the reliable peptide identifications.
2.10. Fluorescence spectroscopy
Fluorescence emission scans were recorded in a Cary Eclipse fluorescence spectrophotometer (Varian Inc., Palo Alto, CA). To eliminate background fluorescence from tyrosine residues, spectra were collected by exciting the sample at 295 nm. Scans were collected at a scan rate of 60 nm/min in a 0.3 cm path length quartz cuvette (Starna Cells, Inc., Atascadero, CA) with the emission slit width set to 5 nm. All scans were collected at room temperature and data plotted in SigmaPlot (Systat, San Jose, CA).
2.11. Circular dichroism (CD) spectroscopy
CD spectra were collected in a Jasco J-815 CD spectrophotometer (Jasco Analytical Instruments, Easton, MD) using a 0.02 cm path length circular quartz cuvette (Hellma USA, Plainview, NY). Samples were scanned from 320 to 190 nm at room temperature. The final spectrum for each sample was an average of eight scans recorded at a scan rate of 50 nm/min with a 0.1 nm step interval and measured in mdeg. Raw scans were processed (subtracted from blank and smoothed) using Spectra Analysis (software provided by Jasco Inc.) and converted to mean residue ellipticity using the following equation: [θ] = 100(signal)/Cnl where, [θ] s the mean residue ellipticity in deg cm2 dmol−1; signal is the raw output in mdeg; C is protein concentration in mM; n is number of residues and l is cell pathlength in cm. The final spectra were plotted in SigmaPlot.
2.12. 2D SDS-PAGE
Purified β2-GPI (190 μg and 170 μg for the human and mouse proteins, respectively) were applied to an 11 cm IPG strip with a pH 3–10 gradient (Bio-Rad), for overnight passive rehydration at 25 °C. Strips were focused at 20 °C with the following program: 20 min with a linear ramp (0–250 V), 2.5 h with a linear ramp (250–8000 V), 20,000 V h with a rapid ramp (8000 V). Once the IEF was completed, the strips were equilibrated in Equilibration Buffer I (6 M urea, 2% SDS, 0.375 Tris–HCl (pH 8.8), 20% glycerol, and 2% (w/v) DTT) (Bio-Rad) followed by another 15 min in Equilibration Buffer II (Equilibration Buffer I containing 2.5% iodoacetamide in place of DTT) (Bio-Rad). 1% agarose solution with 0.01% bromophenol blue was laid on the top of gels to protect the strip. SDS–PAGE was performed using a 10% Criterion Tris–HCl Precast Gel (Bio-Rad). The second dimension was run at 150 V until the bromophenol blue dye front completely migrated out of the bottom of the gel. The gels were stained with Bio-Safe Coomassie Brilliant Blue G-250 (Bio-Rad).
2.13. Anti- 2-GPI ELISA
Polystyrene microtiter plates were coated overnight with 8 μg/mL mouse or human β2-GPI in 0.02 M carbonate buffer, pH 9.6. Purified mouse and human Ig were applied to appropriate wells at 1 mg/mL, with an anti-mouse β2-GPI monoclonal Ab (FC1) (Fleming et al., 2004) and goat anti-human β2-GPI Ab(Bethyl Labs, Montgomery, TX) used as positive controls, for 1 h at room temperature. After washing, appropriate HRP-conjugated secondary Ab (Jackson Immunoresearch) were then applied for 1 h at room temperature and developed using TMB 1-Component substrate (KPL, Gaithersburg, MD).
2.14. Immunoprecipitation
PureProteome Protein G magnetic beads (Millipore) were coated with antibody from C57Bl/6 mice prior to adding 4 μM purified β2-GPI and incubating overnight at 4 °C. The beads were washed and bound β2-GPI was eluted per manufacturer’s directions. Samples were analyzed on 10% SDS-PAGE and Western blotted for β2-GPI using HRP-goat anti-human β2-GPI or anti-mouse IgG antibody.
2.15. Statistics
All data were evaluated by one-way ANOVA with Newman–Keuls post hoc analysis (GraphPad/Instat Software) and are presented as mean ± SEM.
3. Results
3.1. Human but not mouse β2-GPI attenuates IR-induced intestinal injury in a dose dependent manner
Previous studies strongly support the view that β2-GPI is involved in one of the sequential events leading to IR-induced injury (Fleming et al., 2004). In fact, peptides derived from the β2-GPI lipid binding domain (domain 5) inhibit IR-induced complement deposition and subsequent intestinal damage and inflammation (Fleming et al., 2010). Initial ELISA data confirmed the presence of significantly more β2-GPI-Ab complexes within tissues from IR-treated mice (748 ± 97 relative units) than within tissues from Sham treated animals (233 ± 65 relative units). As the β2-GPI-Ab complexes were limited in uninjured tissue, these studies suggested that treatment with exogenous β2-GPI may provide additional antigen for increased Ab binding (Fig. 1B) and could exacerbate intestinal injury and inflammation.
To understand the specific role of β2-GPI in IR-induced tissue damage, we tested the hypothesis by treating wildtype, C57Bl/6 mice with purified human or mouse β2-GPI immediately prior to subjecting the mice to Sham or IR. As expected, IR induced significant injury when compared to Sham treatment (Fig. 2A, B, C). However, mouse β2-GPI derived from either Rag1−/−, C57Bl/6 or Balb/c mice did not significantly alter IR-induced injury (Fig. 2A, E and data not shown). Surprisingly, human β2-GPI significantly inhibited IR-induced intestinal injury (Fig. 2A, F) with a mean injury score of 1.44 ± 0.16 compared to 2.33 ± 0.17 in saline-treated mice. Additional studies demonstrated that human β2-GPI attenuated intestinal injury in a dose dependent manner (Fig. 3A). Administration of either 40 or 200 μg/kg β2-GPI prior to IR significantly decreased injury (Fig. 3A). However, 20 μg/kg human β2-GPI did not attenuate IR-induced intestinal injury. Two different commercial preparations of human β2-GPI showed similar results with IR-induced injury scores of 1.17 ± 0.22 and 0.93 ± 0.23 (Arotec, Wellington, New Zealand and Fitzgerald, Acton, MA, respectively). Thus, human, but not mouse, β2-GPI attenuates IR-induced intestinal injury in a dose dependent manner.
Fig. 2.

Human but not mouse β2-GPI treatment alters IR-induced injury and PGE2 production in C57Bl/6 mice. C57Bl/6 mice were subjected to Sham or IR treatment in the presence or absence of β2-GPI purified from Balb/c (Balb B2), Rag-1−/− (Rag-1−/− B2) or Human (Human B2) plasma. Two hours after initiation of reperfusion, mid-jejunal intestinal sections (75–150 villi per animal) were scored (A) and ex vivo PGE2 (D) production were assessed. Representative H&E stained tissue sections (200×) are provided (B, C, E, F). PGE2 values are represented as pg/mg of intestinal protein. Each bar represents 4–10 animals. * = p ≤ 0.05 compared to Sham + peptide, φ = p ≤ 0.05 compared to I/R treatment animals not receiving peptides.
Fig. 3.
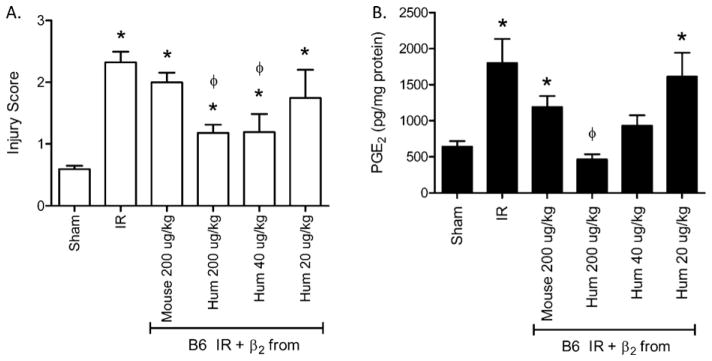
Human β2-GPI attenuates IR-induced intestinal damage and PGE2 production in a dose dependent manner. Mouse β2-GPI (200 μg/kg) or Human β2-GPI (H) at 200, 40 or 20 μg/kg was administered prior to subjecting C57Bl/6 mice to Sham or IR treatment. Two hours after initiation of reperfusion, mid-jejunal intestinal sections (75–150 villi per animal) were scored (A) and ex vivo PGE2 (B) production were assessed. Each bar represents 4–10 animals. * = p ≤ 0.05 compared to Sham, φ = p ≤ 0.05 compared to I/R treatment animals not receiving β2-GPI.
3.2. Human β2-GPI attenuated PGE2 and complement deposition
To examine the mechanism of protection from injury, PGE2 production and complement deposition were analyzed. Similar to injury, intestinal PGE2 production was significantly elevated after IR in the presence or absence of mouse β2-GPI (Fig. 3B). However, only Sham levels of intestinal PGE2 were produced in mice treated with human β2-GPI prior to IR (Fig. 3B). PGE2 production also decreased in a dose dependent manner after administration of human β2-GPI (Fig. 3B). Administering 40 μg/kg human β2-GPI attenuated PGE2 production, and 200 μg/kg prior to IR produced PGE2 concentrations comparable to Sham treatment (Fig. 3B) indicating that 200 μg/kg optimally inhibits both injury and PGE2 production.
As complement is required for IR-induced tissue damage, we examined C3 deposition. Complement deposition was observed in response to IR but not on intestines of Sham treated mice. Similar to PGE2 and injury, C3 deposition significantly decreased after treatment with human β2-GPI when compared to mice subjected to IR only (Fig. 4). Treatment with mouse β2-GPI reduced complement deposition slightly but not to the same extent as treatment with human β2-GPI (Fig. 4). Thus, human β2-GPI attenuated IR-induced injury and inflammation as determined by PGE2 production and complement deposition. Together, these data suggest that human and mouse β2-GPI interact differently in the mouse model of IR-induced tissue damage.
Fig. 4.
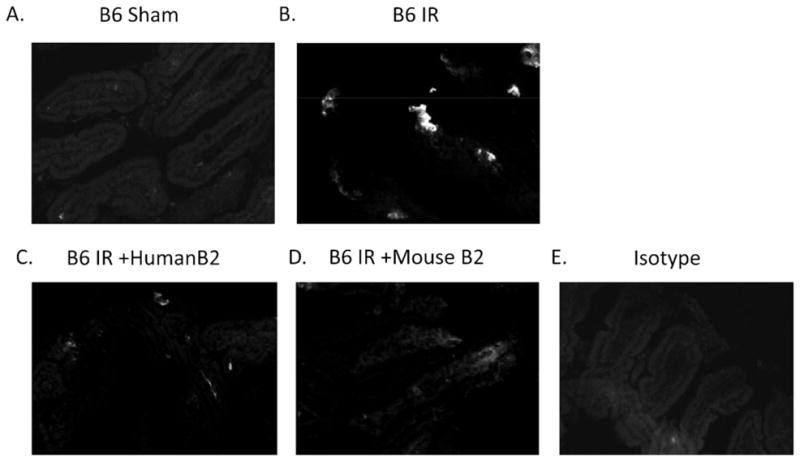
Human β2-GPI attenuates IR-induced complement deposition. C57Bl/6 mice were subjected to Sham or IR treatment in the presence or absence of 200 μg/kg β2-GPI purified from C57Bl/6 (+B6 B2) or Human (+Human B2) plasma. Two hours after initiation of reperfusion, mid-jejunal intestinal sections were collected and stained for C3 deposition. Photomicrographs represent 3 experiments of 5–7 photos per treatment group.
3.3. Human β2-GPI is recognized by mouse antibodies
As Ab are required for complement activation in the current model of IR-induced tissue injury, it is possible that mouse Ab does not recognize surface-bound, human β2-GPI in the same way as endogenous β2-GPI (Fig. 1C). ELISAs performed with purified human and mouse proteins measured the binding of native Ab with each protein. As expected, optimal Ab recognition of β2-GPI was species specific (Fig. 5A). However, Ab purified from mouse plasma clearly recognized bound human β2-GPI as well (open bar). In addition, Ab purified from human plasma recognized bound mouse β2-GPI (black bar). Together these data indicate that mouse Ab are capable of binding surface-bound, human β2-GPI (Fig. 5A) and suggests that the human β2-GPI may compete with mouse β2-GPI for Ab recognition.
Fig. 5.
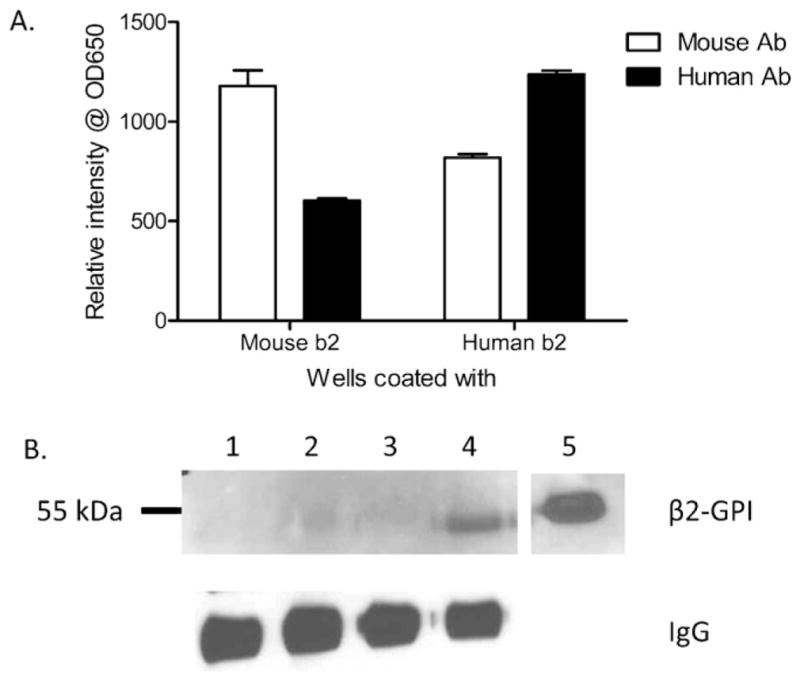
Native mouse antibodies complex with soluble and bound human β2-GPI but only with bound mouse β2-GPI. (A) Mouse (left) or Human (right) purified β2-GPI coated plates were incubated with Ab obtained from naïve C57Bl/6 mice (B6 Ab) or Human sera (human Ab) and developed with TMB to determine optical density. Each bar represents 4 samples and each graph is representative of 3 independent experiments. (B) Protein G purified C57Bl/6 Ab immunoprecipitation of no β2-GPI (lane 1) or 21 ng of soluble Rag-1−/− (lane 2), C57Bl/6 (lane 3), or human (lane 4) β2-GPI was followed by western blot with anti-human β2-GPI (upper panel) or anti-mouse IgG (lower panel). Western blot is representative of 3 immunoprecipitations.
As β2-GPI changes conformation when bound to a surface, it was possible that soluble human and mouse β2-GPI were recognized differently. To determine if soluble human or mouse β2-GPI was bound by mouse Ab similar to bound β2-GPI, we immunoprecipitated the two β2-GPI preparations with mouse Ab. As indicated in Fig. 5B, soluble human β2-GPI bound to beads coated with antibodies from C57Bl/6 mice (lane 4). Surprising, no or very little soluble mouse β2-GPI bound to the mouse anti-β2-GPI-coated beads (lanes 2–3). However, mouse β2-GPI was detectable in the supernatants (data not shown). Equal quantities of Ab were bound to the beads as indicated in the lower panel. Thus, mouse Ab appear to bind soluble human but not mouse β2-GPI, suggesting that administration of human β2-GPI reduces the Ab available for binding the conformationally altered, surface-bound, mouse β2-GPI. Together, these data support the hypothesis illustrated in Fig. 1D.
3.4. Physical and biochemical differences in human and mouse β2-GPI
It is possible that IR-induced injury was attenuated after treatment with human protein due to structural differences in the two proteins that prevented binding of human β2-GPI to the cell surface as suggested in Fig. 1D. We examined the physical and biochemical properties of similarly purified mouse and human β2-GPI. In the presence of thiol reducing agents, SDS-PAGE demonstrated that the two glycoproteins were more than 95% pure with an apparent molecular weight of 58 kDa for the mouse protein and 54 kDa for the human (Supplementary Fig. 1, lanes 1 and 2, respectively). Proteomics analysis confirmed the identity of the two purified proteins, namely mouse (APOH MOUSE) and human (APOH HUMAN) β2-GPI. TOF-MS spectrometry analysis verified the molecular weight difference of the intact proteins. Fig. 6A illustrates the mass difference with a relatively broad peak centered at 48 kDa for mouse β2-GPI (gray line) while the human protein (black line) presents a narrower peak, centered at 45 kDa. Since the MALDI TOF-MS peak width is strictly correlated to the sample heterogeneity, we examined the nature of the glycosylation patterns. The mouse protein was N-glycosylated at Asn residues 162, 183, 193 and 253 (sequence number was followed by registered sequence at Swiss-plot). Human protein possessed a different pattern being N-glycosylated at Asn residues 105, 117, 183 and 193 (Supplementary Fig. 2). PNGase F (Glycopeptidase F) treatment significantly decreased that molecular weights from 58 to 36.5 kDa for the mouse protein and from 54 to 38 kDa for the human, providing further evidence of glycosylation differences between the two species (Supplementary Fig. 1, lanes 3–6). The increased heterogeneity was confirmed by 2D gel electrophoresis which indicated 10 distinct spots with the same molecular weight spanning from pI 5.6 to 7.9 (Fig. 6B). In contrast, the human protein (Fig. 6C) contained only 8 spots ranging from pI 6.0 to 7.5. Thus, despite 76% amino acid identity and 86% sequence similarity, the two proteins possess small but significant differences in heterogeneity.
Fig. 6.

Mouse β2-GPI is different molecular weight and contains additional isoforms of β2-GPI compared to the similarly purified human protein. (A) Molecular weights of purified β2-GPI’s were analyzed by MALDI-TOF in the positive mode using sinapinic acid as a matrix. MS spectra were recorded in linear mode within a mass range from m/z 10,000 to m/z 70,000. The scans for both purified human (black) and mouse (gray) are shown. (B, C) Purified mouse (B) or human (C) β2-GPI were subjected to 2D SDS-PAGE and stained with coomassie blue. Mouse β2-GPI contains additional isoforms of β2-GPI protein compared to the similarly purified human protein. Gels are representative of 2 experimental preparations.
Supplementary material related to this article found, in the online version, at http://dx.doi.org/10.1016/j.molimm.2012.05.018.
To further examine the structural differences between mouse and human β2-GPI, we analyzed the samples using fluorescence spectroscopy and circular dichroism. Fluorescence spectroscopy at 295 nm examined the environment surrounding the tryptophans located at identical positions in the primary sequence. Fig. 7A shows that the fluorescence emission spectrum for each protein is a single peak with a maximum emission at a wavelength of 341 nm for the mouse β2-GPI (dashed line) and at 349 nm for the human protein (solid line). A significant shift towards the lower wavelengths indicates that in the mouse β2-GPI folded structure the tryptophan residues are significantly less solvent exposed compared to the same residues in the human β2-GPI. Circular dichroism confirmed that mouse and human β2-GPI were structurally different with human β2-GPI being less ordered in the far UV region compared to mouse β2-GPI (Fig. 7B). These data provide strong evidence for two distinct conformations for the isolated mouse and human proteins that influence IR-induced injury when administered to wildtype mice.
Fig. 7.

Fluorescence spectroscopy and circular dichroism spectroscopy indicate conformational differences in human and mouse β2-GPI. Purified human (solid line) or mouse (dashed line) β2-GPI were subjected to fluorescence spectroscopy at excitation 295 nm (A) and circular dichroism from 320 to 190 nm (B). Mouse β2-GPI (0.75 mg/mL) and human β2-GPI (0.8066 mg/mL) were normalized for comparison.
4. Discussion
Although the β2-GPI protein was discovered in human plasma 50 years ago, the specific physiological roles remain unknown. β2-GPI does appear to scavenge lipopolysaccharide and inhibit complement activation (Gropp et al., 2011) and thereby plays a role in the innate immune response (Agar et al., 2011b; Gropp et al., 2011). The structural homology is quite conserved among mammalian species and can reach 90–98% sequence identity in specific segments located at the C-terminus where the binding with anionic lipids occurs. Given the homology, most in vivo mouse studies for anti-phospholipid syndrome utilize commercially available human anti-β2-GPI Ab or human β2-GPI (Agostinis et al., 2011; Fleming et al., 2004). We purified human and mouse β2-GPI and compared the biological response of the two proteins in the mouse intestinal IR model. Biochemically, the two species of protein contain significant structural differences. Compared to the human protein, we demonstrated that mouse β2-GPI has an increased molecular weight, increased number of isoforms and differential glycosylation pattern. In addition, the environment surrounding the 5 tryptophan residues, one for each domain, is considerably less solvent accessible in the mouse versus the human protein. Importantly, mouse Ab bound and immunoprecipitated soluble human but not mouse β2-GPI suggesting that the structural differences allow human β2-GPI to sequester mouse anti-β2-GPI Ab and attenuate IR-induced injury and inflammation in the mouse.
Although injury and complement deposition were significantly attenuated after the addition of the human protein, both remained significantly higher than in Sham animals. Complement inhibitors demonstrated a similar degree of attenuation (Rehrig et al., 2001) and suggest that human β2-GPI may contain complement inhibitory activity when employed in mouse models of disease. β2-GPI contains multiple short consensus repeats of the complement control protein family and recent studies demonstrated complement inhibitory effects in vitro. Human β2-GPI contains a C3/C3b binding site which upon binding alters the C3/C3b conformation, allows Factor H binding and inhibits subsequent complement activation (Gropp et al., 2011). This finding supports the current finding in which human β2-GPI attenuates mouse C3 deposition in the mouse model of intestinal IR. While a C3 binding site has not been identified in the mouse protein, it is plausible that the human β2-GPI may be more effective in binding to C3/C3b or that mouse β2-GPI binding to C3/C3b does not expose the Factor H binding site. Either possibility would allow the complement cascade to continue in the presence of mouse but not human β2-GPI.
In contrast to complement deposition and injury, treatment with human β2-GPI inhibited intestinal PGE2 secretion to background levels, suggesting that the pathway to PGE2 production is more sensitive to the conformational changes of β2-GPI than overall injury. The IR-induced production of PGE2 is dependent on both TLR4 and Ab interactions (Moses et al., 2009; Pope et al., 2010; Sparkes et al., 2010). An in vitro study recently demonstrated that human β2-GPI/anti-β2-GPI Ab forms complexes with TLR4 to activate endothelial cells (Allen et al., 2011). Therefore, it is possible that the complex formation of human β2-GPI with mouse Ab is not as efficient in activating endothelial cells to produce PGE2.
It is likely that administration of human but not mouse β2-GPI attenuated IR-induced injury by formation of human β2-GPI/mouse Ab complexes in the serum which reduced Ab binding of endogenous β2-GPI and subsequent injury and PGE2 production. Although mouse Ab do not recognize surface-bound human β2-GPI as effectively as mouse β2-GPI, mouse Ab recognized soluble human β2-GPI significantly better than mouse β2-GPI. These data suggest that human β2-GPI/mouse Ab complexes may indeed form in the mouse. Previous studies also indicated that immunization of mice with human β2-GPI produced Ab which induced fetal resorption presumably by recognition of mouse β2-GPI (Agostinis et al., 2011). Together these data suggest that the human β2-GPI binding to mouse tissue may differ from mouse β2-GPI binding and tissue activation state may be important.
Biochemical analysis of the human and mouse β2-GPI suggest a number of possibilities for the observed biological differences. 2D electrophoresis indicated multiple isoforms, similar to a previous report of commercially available human β2-GPI (Buttari et al., 2005). Although the amino acid composition of β2-GPI indicates that the mouse protein is more basic (theoretical pI = 8.62) than the human protein (theoretical pI = 8.37), 2D gel analysis shows that the average actual pI of the mouse protein is more acidic suggesting that the amino acid differences are compensated by negative charges introduced through post-translational modifications. The human native β2-GPI is decorated with a variable number of sialic acid residues (Kondo et al., 2009; Schousboe, 1983). The addition of variable numbers of sialic acid residues and/or other carbohydrate chains would explain the multiple isoforms found on the 2D gel. Although this explanation is in agreement with previous human protein studies (Gries et al., 1989; Schousboe, 1983), our data do not exclude the possibility that the altered pI is derived from other sources including partial deamination of glutamine or asparagine residues.
Additional biochemical measures, fluorescent spectra and circular dichroism, indicated differences in the folded states of the proteins. The wavelength of the fluorescence emission peak of the mouse protein is significantly lower than that of the human protein, suggesting the tryptophans residues are not exposed in the mouse β2-GPI. In addition, circular dichroism studies on the human protein are reminiscent of previous studies performed with the human β2-GPI, which contain a weak negative at 195 nm (Lee et al., 1983). However, we showed that the mouse β2-GPI contained a significant negative peak centered around 208 nm suggesting a conformational change. Thus, fluorescence, circular dichroism and overall carbohydrate data suggest a different or dynamic structural environment with exposure of different surface residues which are critical to interaction with other proteins. These changes appear to influence the activation of the inflammatory cascade which mediates IR-induced intestinal injury.
Supplementary Material
Acknowledgments
We would like to thank Mr. Andrew Fritze for technical assistance with immunohistochemistry and PGE2 assays. This work was supported by NIH grant AI061691 (SDF) and P20 RR017686 (SDF), and RR016475 (SDF) from the Institutional Development Award Program of the National Center for Research Resources and the Johnson Center for Basic Cancer Research (SDF). Support for the project was also made possible in part by a KSU Targeted Excellence Award (JMT) and an NSF Major Research Instrumentation Program Grant No. 0521587 (JMT).
References
- Agar C, de Groot PG, Marquart JA, Meijers JC. Evolutionary conservation of the lipopolysaccharide binding site of beta2-glycoprotein I. Thrombosis and Haemostasis. 2011a;106:1069–1075. doi: 10.1160/TH11-05-0333. [DOI] [PubMed] [Google Scholar]
- Agar C, de Groot PG, Morgelin M, Monk SD, van Os G, Levels JH, de Laat B, Urbanus RT, Herwald H, van der Poll T, Meijers JC. {beta}2-Glycoprotein I: a novel component of innate immunity. Blood. 2011b;117:6939–6947. doi: 10.1182/blood-2010-12-325951. [DOI] [PubMed] [Google Scholar]
- Agar C, van Os GM, Morgelin M, Sprenger RR, Marquart JA, Urbanus RT, Derk-sen RH, Meijers JC, de Groot PG. {beta}2-Glycoprotein I can exist in two conformations: implications for our understanding of the antiphospholipid syndrome. Blood. 2010;116:1336–1343. doi: 10.1182/blood-2009-12-260976. [DOI] [PubMed] [Google Scholar]
- Agostinis C, Biffi S, Garrovo C, Durigutto P, Lorenzon A, Bek A, Bulla R, Grossi C, Borghi MO, Meroni P, Tedesco F. In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood. 2011;118:4231–4238. doi: 10.1182/blood-2011-01-333617. [DOI] [PubMed] [Google Scholar]
- Allen KL, Fonseca FV, Betapudi V, Willard B, Zhang J, McCrae KR. A novel pathway for human endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood. 2011;119:884–893. doi: 10.1182/blood-2011-03-344671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austen WG, Kyriakides C, Favuzza J, Wang Y, Kobzik L, Moore FD, Hechtman HB. Intestinal ischemia-reperfusion injury is mediated by the membrane attack complex. Surgery. 1999;126:343–348. [PubMed] [Google Scholar]
- Avrameas S. Natural autoantibodies: from ‘horror autotoxicus’ to ‘gnothi seauton’. Immunology Today. 1991;12:154–159. doi: 10.1016/S0167-5699(05)80045-3. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K, Chandra J, Schroit AJ. Immune clearance of phosphatidylserine-expressing cells by phagocytes. The role of beta2-glycoprotein I in macrophage recognition. Journal of Biological Chemistry. 1997;272:31113–31117. doi: 10.1074/jbc.272.49.31113. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K, Maiti SN, Schroit AJ. Recruitment of beta-2-glycoprotein 1 to cell surfaces in extrinsic and intrinsic apoptosis. Apoptosis. 2005;10:439–446. doi: 10.1007/s10495-005-0817-3. [DOI] [PubMed] [Google Scholar]
- Bouma B, de Groot PG, van den Elsen JM, Ravelli RB, Schouten A, Simmelink MJ, Derksen RH, Kroon J, Gros P. Adhesion mechanism of human beta(2)-glycoprotein I to phospholipids based on its crystal structure. EMBO Journal. 1999;18:5166–5174. doi: 10.1093/emboj/18.19.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttari B, Profumo E, Mattei V, Siracusano A, Ortona E, Margutti P, Salvati B, Sorice M, Rigano R. Oxidized beta2-glycoprotein I induces human dendritic cell maturation and promotes a T helper type 1 response. Blood. 2005;106:3880–3887. doi: 10.1182/blood-2005-03-1201. [DOI] [PubMed] [Google Scholar]
- Cabiedes J, Cabral AR, Alarcon-Segovia D. Clinical manifestations of the antiphospholipid syndrome in patients with systemic lupus erythematosus associate more strongly with anti-beta 2-glycoprotein-I than with antiphospholipid antibodies. Journal of Rheumatology. 1995;22:1899–1906. [PubMed] [Google Scholar]
- Chen J, Crispin JC, Dalle Lucca J, Tsokos GC. A novel inhibitor of the alternative pathway of complement attenuates intestinal ischemia/reperfusion-induced injury. Journal of Surgical Research. 2009;167:e131–e136. doi: 10.1016/j.jss.2009.05.041. [DOI] [PubMed] [Google Scholar]
- Chiu CJ, McArdle AH, Brown R, Scott HJ, Gurd FN. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Archives of Surgery. 1970;101:478–483. doi: 10.1001/archsurg.1970.01340280030009. [DOI] [PubMed] [Google Scholar]
- Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, Bossi F, Ziller F, Sblattero D, Meroni P, Tedesco F. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. 2005;106:2340–2346. doi: 10.1182/blood-2005-03-1319. [DOI] [PubMed] [Google Scholar]
- Fleming SD, Egan RP, Chai C, Girardi G, Holers VM, Salmon J, Monestier M, Tsokos GC. Anti-phospholipid antibodies restore mesenteric ischemia/reperfusion-induced injury in complement receptor 2/complement receptor 1-deficient mice. Journal of Immunology. 2004;173:7055–7061. doi: 10.4049/jimmunol.173.11.7055. [DOI] [PubMed] [Google Scholar]
- Fleming SD, Pope MR, Hoffman SM, Moses T, Bukovnik U, Tomich JM, Wagner LM, Woods KM. Domain V peptides inhibit beta2-glycoprotein I-mediated mesenteric ischemia/reperfusion-induced tissue damage and inflammation. Journal of Immunology. 2010;185:6168–6178. doi: 10.4049/jimmunol.1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruchterman TM, Spain DA, Wilson MA, Harris PD, Garrison RN. Complement inhbition prevents gut ischemia and endothelial cell dysfunction after hemorrhage/resuscitation. Surgery. 1998;124:782–792. doi: 10.1067/msy.1998.91489. [DOI] [PubMed] [Google Scholar]
- Gries A, Nimpf J, Wurm H, Kostner GM, Kenner T. Characterization of isoelectric subspecies of asialo-beta 2-glycoprotein I. Biochemical Journal. 1989;260:531–534. doi: 10.1042/bj2600531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallstrom T, Skerka C. beta-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. 2011;118:2774–2783. doi: 10.1182/blood-2011-02-339564. [DOI] [PubMed] [Google Scholar]
- Hagihara Y, Hong DP, Hoshino M, Enjyoji K, Kato H, Goto Y. Aggregation of beta(2)-glycoprotein I induced by sodium lauryl sulfate and lysophospholipids. Biochemistry. 2002;41:1020–1026. doi: 10.1021/bi015693q. [DOI] [PubMed] [Google Scholar]
- Hammel M, Kriechbaum M, Gries A, Kostner GM, Laggner P, Prassl R. Solution structure of human and bovine beta(2)-glycoprotein I revealed by small-angle X-ray scattering. Journal of Molecular Biology. 2002;321:85–97. doi: 10.1016/s0022-2836(02)00621-6. [DOI] [PubMed] [Google Scholar]
- Hart ML, Ceonzo KA, Shaffer LA, Takahashi K, Rother RP, Reenstra WR, Buras JA, Stahl GL. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. Journal of Immunology. 2005;174:6373–6380. doi: 10.4049/jimmunol.174.10.6373. [DOI] [PubMed] [Google Scholar]
- Hill J, Lindsay TF, Ortiz F, Yeh CG, Hechtman HB, Moore FD. Soluble complement receptor type 1 ameliorates the local and remote organ injury after intestinal ischemia-reperfusion in the rat. Journal of Immunology. 1992;149:1723–1728. [PubMed] [Google Scholar]
- Kondo A, Miyamoto T, Yonekawa O, Giessing AM, Osterlund EC, Jensen ON. Glycopeptide profiling of beta-2-glycoprotein I by mass spectrometry reveals attenuated sialylation in patients with antiphospholipid syndrome. Journal of Proteomics. 2009;73:123–133. doi: 10.1016/j.jprot.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, Andrews KA, Markaryan A, Quigg RJ, Silverman GJ, Tsokos GC, Holers VM. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. Journal of Immunology. 2009;182:5363–5373. doi: 10.4049/jimmunol.0803980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante P, Amireault P, Subang R, Dieude M, Levine JS, Rauch J. Interaction of beta2-glycoprotein I with lipopolysaccharide leads to toll-like receptor 4 (TLR4)-dependent activation of macrophages. Journal of Biological Chemistry. 2011;286:42494–42503. doi: 10.1074/jbc.M111.230383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NS, Brewer HB, Jr, Osborne JC., Jr beta 2-Glycoprotein I. Molecular properties of an unusual apolipoprotein, apolipoprotein H. Journal of Biological Chemistry. 1983;258:4765–4770. [PubMed] [Google Scholar]
- Manfredi AA, Rovere P, Heltai S, Galati G, Nebbia G, Tincani A, Balestrieri G, Sabbadini MG. Apoptotic cell clearance in systemic lupus erythematosus. II. Role of beta2-glycoprotein I. Arthritis and Rheumatism. 1998;41:215–223. doi: 10.1002/1529-0131(199802)41:2<215::AID-ART5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a natural immune memory. Immunological Reviews. 2000;175:70–79. [PubMed] [Google Scholar]
- Meroni PL, Raschi E, Testoni C, Tincani A, Balestrieri G, Molteni R, Khamashta MA, Tremoli E, Camera M. Statins prevent endothelial cell activation induced by antiphospholipid (anti-beta2-glycoprotein I) antibodies: effect on the proadhesive and proinflammatory phenotype. Arthritis and Rheumatism. 2001;44:2870–2878. doi: 10.1002/1529-0131(200112)44:12<2870::aid-art475>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Moses T, Wagner L, Fleming SD. TLR4-mediated Cox-2 expression increases intestinal ischemia/reperfusion-induced damage. Journal of Leukocyte Biology. 2009;86:971–980. doi: 10.1189/jlb.0708396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Wasylik S, Morgelin M, Olin AI, Meijers JC, Derksen RH, de Groot PG, Herwald H. The antibacterial activity of peptides derived from human beta-2 glycoprotein I is inhibited by protein H and M1 protein from Streptococcus pyogenes. Molecular Microbiology. 2008;67:482–492. doi: 10.1111/j.1365-2958.2007.05974.x. [DOI] [PubMed] [Google Scholar]
- Pope MR, Hoffman SM, Tomlinson S, Fleming SD. Complement regulates TLR4-mediated inflammatory responses during intestinal ischemia reperfusion. Molecular Immunology. 2010;48:356–364. doi: 10.1016/j.molimm.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehrig S, Fleming SD, Anderson J, Guthridge JM, Rakstang J, McQueen CE, Holers VM, Tsokos GC, Shea-Donohue T. Complement inhibitor, complement receptor 1-related gene/protein y-Ig attenuates intestinal damage after the onset of mesenteric ischemia/reperfusion injury in mice. Journal of Immunology. 2001;167:5921–5927. doi: 10.4049/jimmunol.167.10.5921. [DOI] [PubMed] [Google Scholar]
- Schousboe I. Characterization of subfractions of beta 2-glycoprotein I: evidence for sialic acid microheterogeneity. International Journal of Biochemistry. 1983;15:35–44. doi: 10.1016/0020-711x(83)90008-3. [DOI] [PubMed] [Google Scholar]
- Schultze HE. Glycoproteins of human plasma. Bulletin der Schweizerischen Akademie der Medizinischen Wissenschaften. 1961;17:77–91. [PubMed] [Google Scholar]
- Schwarzenbacher R, Zeth K, Diederichs K, Gries A, Kostner GM, Laggner P, Prassl R. Crystal structure of human beta2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO Journal. 1999;18:6228–6239. doi: 10.1093/emboj/18.22.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren RW, Colleton C, Shea-Donohue TS. Intestinal myoelectric response in two different models of acute enteric inflammation. American Journal of Physiology. 1994;267:G329–G337. doi: 10.1152/ajpgi.1994.267.3.G329. [DOI] [PubMed] [Google Scholar]
- Sparkes BL, Slone EE, Roth M, Welti R, Fleming SD. Intestinal lipid alterations occur prior to antibody-induced prostaglandin E2 production in a mouse model of ischemia/reperfusion. Biochimica et Biophysica Acta. 2010;1801:517–525. doi: 10.1016/j.bbalip.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefas I, Dubois G, Tigrett S, Lucarz E, Veas F. Apolipoprotein H, an acute phase protein: a performing tool for ultra-sensitive detection and isolation of microorganisms from different origins. In: Veas F, editor. InTechWeb. 2011. [Google Scholar]
- Tolomeo T, Rico De Souza A, Roter E, Dieude M, Amireault P, Subang R, Levine JS, Rauch J. T cells demonstrate a Th1-biased response to native beta2-glycoprotein I in a murine model of anti-phospholipid antibody induction. Autoimmunity. 2009;42:292–295. doi: 10.1080/08916930902828254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser MR, Williams JP, Moore FD, Kobzik L, Ma M, Hechtman HB, Car-roll MC. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. Journal of Experimental Medicine. 1996;183:2343–2348. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JP, Pechet TTV, Weiser MR, Reid R, Kobzik L, Moore FD, Carroll MC, Hechtman HB. Intestinal reperfusion injury is mediated by IgM and complement. Journal of Applied Physiology. 1999;86:938–942. doi: 10.1152/jappl.1999.86.3.938. [DOI] [PubMed] [Google Scholar]
- Wurm H. beta 2-Glycoprotein-I (apolipoprotein H) interactions with phospholipid vesicles. International Journal of Biochemistry. 1984;16:511–515. doi: 10.1016/0020-711x(84)90168-x. [DOI] [PubMed] [Google Scholar]
- Zhang M, Austen WG, Chiu I, Alicot EM, Humg R, Ma M, Verna N, Xu M, Hechtman HB, Moore FD, Carroll MC. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3886–3891. doi: 10.1073/pnas.0400347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.