

Abstract
Zinc finger nucleases (ZFNs) have become powerful tools to deliver a targeted double-strand break at a pre-determined chromosomal locus in order to insert an exogenous transgene by homology-directed repair. ZFN-mediated gene targeting was used to generate both single-allele chemokine (C-C motif) receptor 5 (CCR5)-modified human induced pluripotent stem cells (hiPSCs) and biallele CCR5-modified hiPSCs from human lung fibroblasts (IMR90 cells) and human primary cord blood mononuclear cells (CBMNCs) by site-specific insertion of stem cell transcription factor genes flanked by LoxP sites into the endogenous CCR5 locus. The Oct4 and Sox2 reprogramming factors, in combination with valproic acid, induced reprogramming of human lung fibroblasts to form CCR5-modified hiPSCs, while 5 factors, Oct4/Sox2/Klf4/Lin28/Nanog, induced reprogramming of CBMNCs. Subsequent Cre recombinase treatment of the CCR5-modified IMR90 hiPSCs resulted in the removal of the Oct4 and Sox2 transgenes. Further genetic engineering of the single-allele CCR5-modified IMR90 hiPSCs was achieved by site-specific addition of the large CFTR transcription unit to the remaining CCR5 wild-type allele, using CCR5-specific ZFNs and a donor construct containing tdTomato and CFTR transgenes flanked by CCR5 homology arms. CFTR was expressed efficiently from the endogenous CCR5 locus of the CCR5-modified tdTomato/CFTR hiPSCs. These results suggest that it might be feasible to use ZFN-evoked strategies to (1) generate precisely targeted genetically well-defined patient-specific hiPSCs, and (2) then to reshape their function by targeted addition and expression of therapeutic genes from the CCR5 chromosomal locus for autologous cell-based transgene-correction therapy to treat various recessive monogenic human diseases in the future.
Introduction
The creation of designer zinc finger nucleases (ZFNs) and the development of ZFN-mediated gene targeting has conferred molecular biologists with the ability to site specifically and permanently modify plant and mammalian genomes, including the human genome, via homology-directed repair of a targeted genomic double-strand break (DSB) [1–4]. Our lab at the Johns Hopkins Medical Institutions pioneered the work on the creation of designer ZFNs [1], and then in collaboration with Dana Carroll's lab in Utah, showed the utility of ZFNs in gene targeting using frog oocytes as a model system [2]. Published reports from several labs have now firmly established ZFN-mediated gene targeting as a powerful research tool for site-specific and precise modification of the human genome. Site-specific modification of the human genome using designed ZFNs [and more recently using transcription activator like effector nucleases (TALENs)], has been successfully demonstrated in a variety of human cells and other cell types [5–24]. High rate of endogenous gene modification efficiencies (>10%) have been achieved using ZFN-driven approach [8]. Several labs have also reported successful ZFN-mediated gene targeting of human stem progenitor cells (HSPCs) [22–25], human induced pluripotent stem cells (hiPSCs), and human embryonic stem cells (hESCs) [14–19]. High targeting efficiency (∼50% of clones selected with antibiotics) in hESCs and iPSCs, both at expressed and silent genes, was observed in these studies.
In the present study, we investigate the potential of ZFN-evoked strategies to generate precisely targeted genetically well-defined hiPSCs, and then to reshape the functionality of such hiPSCs (Fig. 1). Retro-virally based methods that are often used for the generation hiPSCs could lead to insertional mutagenesis and oncogene activation, and the resulting hiPSCs may not be suitable for clinical studies. Here, we demonstrate the use of ZFN technology for the generation of precisely targeted genetically well-defined chemokine (C-C motif) receptor 5 (CCR5)-modified hiPSCs (both single-allele CCR5-modified hiPSCs and biallele CCR5-modified hiPSCs) by site-specific addition of Oct4/Sox2 genes to the CCR5 locus of human lung fibroblasts, in conjunction with small molecule HDAC inhibitor, valproic acid (VPA). The stem cell factor genes are then removed by treatment of CCR5-modified hiPSCs with Cre recombinase. We then demonstrate further genetic engineering of the single-allele CCR5-modified hiPSCs generated above by targeted addition and expression of a therapeutic gene (the large CFTR transcription unit in this case) at the remaining wild-type CCR5 allele. We also demonstrate the use of ZFN technology for the generation of precisely targeted, genetically well-defined CCR5-modified hiPSCs (both single-allele CCR5-modified hiPSCs and biallele CCR5-modified hiPSCs) by site-specific addition of Oct4/Sox2/Klf4/Lin28/Nanog genes to the CCR5 locus of human primary cord blood mononuclear cells (CBMNCs).
FIG. 1.

Schematic diagram showing generation of single-allele chemokine (C-C motif) receptor 5 (CCR5)-modified human induced pluripotent stem cells (hiPSCs) (ii & iv) and biallele CCR5-modified hiPSCs (iii & v) by targeted insertion of stem cell factor genes (i) at the CCR5 locus of the human lung fibroblasts and further genetic engineering of single-allele CCR5-modified hiPSCs (after Cre recombinase treatment) (iv) by targeted addition and expression of CFTR transcription unit (vi) from the remaining wild-type CCR5 allele, using CCR5-specific zinc finger nucleases (ZFNs). pA denotes polyA sequence. Color images available online at www.liebertpub.com/scd
We chose to target the CCR5 locus of the human genome in our study [23]. CCR5 is a co-receptor involved in HIV-1 infection of macrophages and T cells. Homozygous inactivating mutations of CCR5 are present in a subset of healthy humans, and CCR5 is thought to be dispensable for normal cellular differentiation and function. Importantly, ZFN-mediated inactivating mutation of CCR5 should not affect the biology of human hiPSCs and could potentially serve as a safe harbor locus for targeted addition of a therapeutic gene for functional protein complementation in cells with the corresponding recessive monogenic defects. Previous reports have shown that CCR5 could potentially be an attractive therapeutic target for future gene therapy for treatment and/or prevention of HIV/AIDS [22–25].
Materials and Methods
The 4-finger CCR5-specific ZFPs fused to wild-type FokI cleavage domain or to FokI obligate heterodimer variants were originally reported by Lombardo et al. [22,26,27]. The CCR5-specific ZFNs used in this study were generated by fusing the above 4-finger ZFPs to the FokI nuclease domain heterodimer variant pair (REL_DKK), which is described elsewhere [4]. The nucleotide sequences of the polymerase chain reaction (PCR) primers and PCR amplification conditions used in this study are shown in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/scd).
Construction of donor plasmids containing 2 and 5 reprogramming genes
The open reading frames (ORFs) of the human Oct4 and Sox2 transcription factors were amplified from cDNA clones obtained from Harvard Proteomics. The PCR-amplified DNAs were placed between Foot and Mouth disease virus 2A sequences to allow efficient polycistronic expression [28]. A single CMV promoter, which drives the expression of these 2 transgenes, along with marker genes (either enhanced green fluorescence protein (eGFP) or puromycin resistance gene) were cloned into pNTKV using AflII and BamHI restriction sites. The transgene cassette was flanked by loxP sites for Cre-mediated excision of the transgenic cassette. This whole transgene cassette was flanked by 750 bp of endogeneous CCR5 locus sequence on both sides for ZFN-evoked homology-directed repair. Both the CCR5 homology arms were PCR amplified using specific primers and cloned in pNTKV vector using restriction enzymes AscI and HpaI (750 bp of left homology arm) and HpaI and AscI (750 bp of right homology arm). The final plasmids were named as pPBPL-Oct4/Sox2/eGFP and pPBPL-Oct4/Sox2/eGFP/PuroR. Using a similar protocol, a donor plasmid containing 5 stem cell factor genes, Oct4/Sox2/Klf4/Lin28/Nanog/eGFP, was constructed.
Human cell culture
Human lung fibroblast IMR90 cells obtained from ATCC were cultured in minimum essential medium (Quality Biological, Inc.) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 0.1 mM non-essential amino acids (Invitrogen), and 1.0 mM sodium pyruvate (Invitrogen). hiPSCs were maintained on irradiated mouse embryonic fibroblasts (iMEF) (R&D Systems) in Dulbecco's modified Eagle's medium/nutrient F12 ham (DMEM/F12) culture medium supplemented with 20% knock-out serum replacer, 0.1 mM non-essential amino acids, 1 mM l-glutamine, 0.1 mM β-mercaptoethanol, and 100 ng/mL human basic fibroblast growth factor (all from Invtrogen).
Plasmid transfection and reprogramming
IMR90 cells were seeded at 3×105 cells per well of 6-well plates (day 0) in IMR90 growth medium. On day 1, cells were co-transfected with plasmids of 2 ZFNs (pPBPL-REL and pPBPL-DKK), and the donor transgene plasmid (either pPBPL-Oct4/Sox2/eGFP or pPBPL-Oct4/Sox2/eGFP/PuroR), which were introduced with 1:3 ratio using TransIT transfection reagent (Mirus). The transfection was repeated on day 3 using the same plasmids. On day 5, cells were digested off the culture plate with 0.05% trypsin-ethylenediaminetetraacetic acid (Invitrogen). Cells were then transferred onto an iMEF feeder layer in a gelatin-coated 6-well plate and cultured with hESC growth medium. hiPSC colonies with morphology similar to hESC colonies appeared on day 28–30 and were picked and expanded using iMEF feeder layer conditions.
Alkaline phosphatase staining of hiPSCs
Alkaline phosphatase staining was performed using the Vector Red Alkaline phophatase substrate kit I (Vector Laboratories) following the manufacturer's instruction. Briefly, cells were fixed with 2% formaldehyde for 30 min at room temperature (RT) and the colonies were stained with Vector Red substrate working solution for 30 min at RT. After 30 min, wells were rinsed with 100 mM Tris-Cl (pH 8.0) twice and then further rinsed with double distilled water. Cells were viewed under bright field and fluorescence microscope.
Immunostaining of hiPSCs
In order to characterize single-cell colony hiPSCs by immunostaining with various pluripotency markers, cultured hiPSCs were fixed with 100% ice-cold methanol for 20 min, permeabilized with 0.1% Triton X-100 in phosphate buffered saline (PBS) for 20 min, and then washed with PBS for 3 times. The fixed samples were blocked with 0.5% goat serum in PBS for 1 h and incubated with primary antibodies with 0.25% Triton X-100 in PBS for overnight at 4°C. The primary antibodies for Oct3/4 (Stemgent), Sox2 (Stemgent), Nanog, and Tran-1-60 (Applied StemCell, Inc.) were used in the staining. After washing 3 times with PBS, cells were incubated with AlexaFluor-conjugated secondary antibodies (Invitrogen-Molecular Probes) with 2 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI) (Roche) at room temperature in the dark for 1 h.
Removal of Oct4/Sox2/eGFP cassette by transient Cre-recombinase expression
hiPSCs were harvested using accutase (Sigma), and 2×106 cells were resuspended in 100 μL Amaxa mouse ES nucleofection buffer with 3 μg of pNTKV-Cre and transfected using the Nuclecofector II with program A-23. Cells were subsequently plated on iMEF feeder layers in hESC medium supplemented with 10 μM of ROCK inhibitor for the first 24 h. Three days after nucleofection, GFP-negative cells were fluorescence-activated cell sorting (FACS) sorted and collected as single-cell/well in Matrigel 96-well plates and allowed to grow in ROCK inhibitor containing hES medium.
Genotype analysis of CCR5 gene-targeted hiPSC lines
In order to provide the evidence for successful homologous recombination (HR) of donor DNA at the targeted CCR5 chromosomal locus, we performed PCR analysis at the 5′ and 3′ junctions of the donor insertion sites using the corresponding locus-specific primers (Supplementary Table S1). PCR products were amplified from each individual single-cell colony hiPSC clones, subcloned into Escherichia coli, and then the recombinant plasmids sequenced.
Quantitative reverse transcription-polymerase chain reaction analyses
Total RNA was isolated from the hiPSC lines using the RNeasy Mini kit (Qiagen) and treated with Turbo DNA-free kit (Ambion) to remove genomic DNA contamination. One microgram of total RNA was used for the reverse transcription using qScript cDNA superMix (Quanta Biosciences) according to the manufacturer's instructions. Quantitative PCR was performed using TaqMan gene expression assays from Applied Biosystems (ABI). Expression levels of individual transcripts (Oct4, Sox2, Nanog and Rexi) were normalized to glyceraldehyde 3-phosphate dehydrogenase expression.
Construction of CFTR/tdTomato donor plasmid
The CFTR/tdTomato donor plasmid was generated using Gibson isothermal assembly method [29]. The CMV promoter, tdTomato, and polyA were amplified with unique 40 bp overlaps. The ORF of the human CFTR was amplified as 3 pieces of 1.5 kb in size and with unique 40 bp overlaps between adjoining partners from wild-type (WT) cDNA clone. The CAG promoter, which drives the expression of CFTR, was also amplified with unique overlaps. All the PCR products were column purified and Gibson isothermal assembly was used to assemble the target insert in the desired order of fragments onto a pUC19 vector [29]. The assembled product was digested with MfeI and cloned into pNTKV vector, which contained the CCR5 homology arms. The donor construct contained tdTomato gene under the control of the Pcmv promoter, while CFTR cDNA was under the control of Pcag promoter. This whole transgene cassette was flanked by 750 bp of endogeneous CCR5 locus sequence on both sides (the same homology arms that were used in donors containing stem cell factor genes) for ZFN-evoked homology-directed repair.
Southern blot analysis
Genomic DNA isolated from hiPSC lines, after digestion with appropriate restriction enzymes, were resolved on a 0.8% agarose gel and then transferred to a Hybond N+ nylon membrane (GE Healthcare) by capillary transfer method [30]. After transfer and UV cross-linking (Hoefer), the nylon membrane was incubated in prehybridization solution [6× saline sodium citrate (SSC), 5× Denhardts solution, 0.5% sodium dodecyl sulfate (SDS), 100 μg/mL denatured Salmon sperm DNA, and 50% deionized formamide] in rotating incubator for 3 h at 42°C. The prehybridization solution was replaced with hybridization solution (6× SSC, 0.5% SDS, denatured salmon sperm DNA, 50% deionized formamide) and then the appropriate denatured radioactive labeled probe was added to the solution. Hybridization was carried out at 42°C overnight with gentle rotation. The membrane was sequentially washed in 1× SSC, 0.1% SDS, and 0.5× SSC, 0.1% SDS at room temperature for 15 min and exposed to X-ray film with intensifying screens.
Western blot analysis of CFTR expression in CCR5 gene-targeted hiPSC lines
CCR5-CFTR gene-targeted hiPSCs grown in Matrigel 6-well plates were lysed in NP-40 lysis buffer (50 mM Tris-HCl pH 8.0,150 mM NaCl, 1% NP-40, phenlymethyl sulfonyl fluoride (PMSF) 50 μL/mL). Fifty micrograms of total protein of cell extracts was separated on 8% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membrane (Bio-rad Laboratories). The blot was blocked with Tris-buffered saline (TBS)-Tween (0.1% Tween-20 in TBS) supplemented with 5% low fat milk powder (w/v) for 1 h at room temperature and incubated with an Ab217 primary antibody (1:2,000) (UNC Cystic Fibrosis Center). After washing, the membrane was incubated with Peroxidase-AffiniPure Goat Anti-Mouse immunoglobulin G (H+L) secondary antibody (1:2,000) (Jackson ImmunoResearch) for 1 h at room temperature. The membrane was washed again with TBS-Tween (0.1%) and developed using an ECL chemiluminescence detection system (Amersham Biosciences) according to manufacturer's instructions.
Teratoma formation and karyotyping of CFTR-expressing hiPSCs
CFTR-expressing hiPSCs were grown on Matrigel plates were harvested by dispase treatment (1.5 mg/mL). hiPSC aggregates were collected by centrifugation and resuspended in a ratio of 106 cells in 200 μL of hiPSC culture media and injected subcutaneously into NOD-SCID mice. Tumors were observed typically 6–8 weeks after injection. Tumor samples were collected typically in 8–10 weeks after injection. Teratomas were isolated after sacrificing the mice and fixed in formalin. After sectioning, slides containing various regions of teratomas were stained by hematoxylin and eosin. The karyotyping assays using CFTR-expressing hiPSCs were performed by Cell Line Genetics, Inc.
Reprogramming of CBMNCs
Human primary CBMNCs were purchased from Allcells, Inc. The frozen CBMNCs were thawed and then cultured in the serum free medium (SFM) supplemented with cytokines and a hormone: stem cell factor (SCF) (50 ng/mL), interleukin-3 (10 ng/mL), erythropoietin (EPO) (2 U/mL), insulin like growth factor-1 (IGF-1) (40 ng/mL) (all from R&D Systems) and dexamethosone (1 μM; Sigma) [31]. The media were changed at day 3 and 6. Once the overt sign of cell division was observed on day 9, the cells were harvested and 2×106 cells were nucleofected with 1 μg of each ZFN plasmids (pPBPL-REL and pPBPL-DKK) and 8 μg donor plasmid (pNTKV-5F) using nuclofector II with program T-016. The nucleofected CBMNCs were cultured in the SFM for 2 days and then transferred to iMEF feeder cells for reprogramming.
Results
A schematic diagram of the experimental strategy used in this study to generate hiPSCs and then to reshape their functionality using CCR5-specific ZFNs is shown in Fig. 1. In the first step, we generated both single-allele CCR5-modified hiPSCs [Fig. 1(ii)] and biallele CCR5-modified hiPSCs [Fig. 1(iii)] by targeted addition of stem cell factor genes flanked by loxP sites [Fig. 1(i)] at the CCR5 locus of human lung fibroblasts. Cre treatment of the reprogrammed cells allowed removal of the stem cell factor genes [Fig. 1(iv, v)]. In the second step, we used CCR5-specific ZFNs for further genetic engineering of the single-allele CCR5-modified hiPSCs by targeted addition and expression of the large CFTR transcription unit (using the remaining wild-type CCR5 allele) [Fig. 1(vi)].
Generation of hiPSCs by targeted insertion of Oct4/Sox2 genes at the CCR5 locus of human lung fibroblasts, using CCR5-specific ZFNs
We generated CCR5-modified hiPSCs by targeted insertion of 2 reprogramming genes (Oct4/Sox2) flanked by loxP sites, at the CCR5 locus of human lung fibroblasts (IMR90 cells), using ZFN-mediated gene targeting in combination with small molecule HDAC inhibitor, VPA (Figs. 2 and 3). The donor plasmid contained a single polycistronic cassette in which Oct4, Sox2, and eGFP (and/or Puro) were each separated by 2A peptides, as well as the Pcmv promoter flanked by CCR5 homology regions (Fig. 1) for targeted insertion at the CCR5 locus of human lung fibroblasts. Twenty individual single-cell colonies with morphology similar to hES cells were picked, grown, and characterized further for genotype of the CCR5 locus by PCR and sequencing. Complete data for 4 single-allele and biallele CCR5-modified IMR90 hiPSCs are shown in Figs. 2 and 3, respectively. The CCR5-gene-modified hiPSC single-cell colonies were characterized for expression of pluripotency markers before Cre treatment [Figs. 2(i) and 3(i)]. As expected, all the pluripotency markers were expressed in all the clones that were examined. PCR analysis of the 5′ and 3′ junctions of the donor insertion site [using one of the primers anchored outside the CCR5 homology arms of the donor and the other anchored within the donor as shown in Fig. 2(ii) for single-allele CCR5-modified hiPSCs; and as shown in Fig. 3(ii) for biallele CCR5-modified hiPSCs] yielded the expected 1.8 and 2.7 kb fragments respectively [Figs. 2(iii, iv) and 3(iii, iv)] indicating donor insertion at the CCR5 locus. The PCR fragments were then cloned and sequenced to determine the nucleotide sequence at the 5′ and 3′ junctions of the donor insertion site in the CCR5-modified hiPSCs. The sequencing data further confirmed the presence of both single-allele CCR5-modified hiPSCs resulting from HR (Fig. 2; Supplementary Table S2a) and biallele CCR5-modified hiPSCs resulting from HR and nonhomologous end joining (NHEJ) (Fig. 3; Supplementary Fig. S1a; Supplementary Tables S2a and S3). Although ZFN-mediated gene targeting resulted in highly specific integration of the pluripotency genes into the targeted CCR5 locus of the human lung fibroblasts, the reprogramming, however, occurred at low efficiency (Supplementary Table S4a, b; ∼120 hiPSC colonies per 300,000 treated cells with GFP donor and ∼70 hiPSC colonies per 300,000 treated cells with puromycin donor). The ratio of single-allele (16 single-cell colonies) versus biallele (4 single-cell colonies) CCR5-modified colonies for IMR90-derived hiPSC lines was 4:1 (Supplementary Table S4b).
FIG. 2.

Generation of CCR5-modified hiPSCs from human lung fibroblasts (IMR90 cells) by targeted insertion of Oct4/Sox2 transcription factors at the CCR5 locus using designed ZFNs, in conjunction with small molecule inhibitor, valproic acid. (i) Morphology of single-allele CCR5-modified hiPSCs, before Cre treatment. (A, B) Bright field images of the morphology of 2 single-allele CCR5-modified hiPSC lines. (C, D) eGFP fluorescence images of the hiPSC lines shown in (A, B). (E, F), alkaline phosphatase staining of 2 single-allele CCR5-modified hiPSC lines. Immunostaining for Oct4/Sox2/Nanog/Tra1-60 and DAPI staining of single-allele CCR5-modified hiPSCs are also shown. (ii) Schematic representation of donor (Oct4/Sox2/eGFP flanked by CCR5 homology arms) insertion site at the CCR5 locus of single-allele CCR5-modified hiPSCs. Polymerase chain reaction (PCR) primers anchored outside the CCR5 homology arms and primers anchored inside the donor for the 5′ and 3′ junction sites are shown. pA denotes polyA sequence. (iii) PCR analysis of 5′ junction of donor insertion site in 4 different CCR5-modified hiPSC single-cell colonies, before Cre treatment. Lanes: 1–4, single-allele CCR5-modified hiPSC lines; 5, Control IMR90 cells; and 6, 1 kb ladder. PCR analysis yielded the expected band size (1.8 kb) confirming insertion of the donor at the CCR5 locus. (iv) PCR analysis of 3′ junction of donor insertion site in 4 different CCR5-modified hiPSC single-cell colonies, before CRE treatment. Lanes: 1–4, single-allele CCR5-modified hiPSC colonies; 5, control IMR90 cells; and 6, 1 kb ladder. PCR analysis yields the expected band size (2.7 kb) confirming insertion of the donor at the CCR5 locus. (v) Morphology of single-allele CCR5-modified hiPSCs, post Cre treatment. (A) Bright field images of the morphology 3 representative single-allele CCR5-modified hiPSC lines, post Cre treatment. (B) GFP fluorescence images of single-allele CCR5-modified hiPSC lines shown in (A). Immunostaining for Oct4/Sox2/Nanog/Tra1-60 and DAPI staining of the single-allele CCR5-modified hiPSC lines are also shown. (vi) Southern blot profiles of 4 different single-allele and biallele CCR5-modified IMR90 hiPSC lines, respectively. Genomic DNA was digested with BamHI and hybridized using either 32P-radiolabeled GFP gene or 32P-radiolabeled Sox2 gene. Lanes: 1, IMR90 cells; 2–5, 4 different single-allele CCR5-modified hiPSC lines; 6–9, 4 different biallele CCR5-modified hiPSC lines. The data confirms single insertion of the donor at the targeted CCR5 locus. Target denotes donor (containing Sox2 gene) insertion at the CCR5 locus; Wt denotes endogenous Sox2 genes. Color images available online at www.liebertpub.com/scd
FIG. 3.

Characterization of biallele CCR5-modified hiPSCs, generated from human fibroblasts by targeted insertion of Oct4/Sox2 transcription factors at the CCR5 locus, using CCR5-specific ZFNs. (i) Morphology of biallele CCR5-modified hiPSCs, before CRE treatment. (A, B) Bright field images of the morphology 2 representative biallele CCR5-modified hiPSC colonies. (C, D) eGFP fluorescence images of hiPSC single-cell colonies shown in (A, B). (E, F) Alkaline phosphatase staining of 2 biallele CCR5-modified hiPSC lines. Immunostaining for Oct4/Sox2/Nanog/Tra1-60 and DAPI staining of biallele CCR5-modified hiPSCs are shown. (ii) Schematic representation of donor (Oct4/Sox2/eGFP flanked by CCR5 homology arms) insertion site at the CCR5 locus of single-allele CCR5-modified hiPSCs. PCR primers anchored outside the CCR5 homology arms and primers anchored inside the donor for 5′ and 3′ junction sites are shown. pA denotes polyA sequence. (iii) PCR analysis of 5′ junction of donor insertion site in 4 different biallele CCR5-modified hiPSC lines, before CRE treatment. Lanes: 1–4, biallele CCR5-modified hiPSC lines; 5, control IMR90 cells; and 6, 1 kb ladder. PCR analysis yields the expected band size (1.8 kb) confirming insertion of the donor at the CCR5 locus. (iv) PCR analysis of 3′ junction of donor insertion site in 4 different biallele CCR5-modified hiPSC single-cell colonies, before CRE treatment. Lanes: 1–4, single-allele CCR5-modified hiPSC colonies; 5, control IMR90 cells; and 6, 1 kb ladder. PCR analysis yields the expected band size (2.7 kb) confirming insertion of the donor at the CCR5 locus. (v) Morphology of biallele CCR5-modified hiPSCs, post Cre treatment. (A) Bright field images of the morphology 3 representative biallele CCR5-modified hiPSC lines generated, post Cre treatment. (B) eGFP fluorescence images of the IMR90 hiPSC lines shown in A. Immunostaining for Oct4/Sox2/Nanog/Tra1-60 and DAPI staining of the hiPSC lines are also shown. Color images available online at www.liebertpub.com/scd
The single-allele CCR5-modified hiPSCs and biallele CCR5-modified hiPSCs, respectively, were then treated with Cre recombinase to remove the reprogramming genes (Oct4/Sox2 and eGFP) [Figs. 2(v) and 3(v); Supplementary Fig. S2a]. The Cre-treated CCR5-modified hiPSCs were then characterized for pluripotency markers. As expected, all the pluripotency markers were expressed in all hiPSC clones that were examined [Figs. 2(v) and 3(v)]. PCR analysis of single-cell colonies of the single-allele CCR5-modified hiPSCs and biallele CCR5-modified hiPSCs were performed using similar primers flanking the CCR5-specific ZFN target sites, which yielded the expected closely spaced (∼0.9 kb size) DNA fragments (Fig. 4). The PCR results for single-allele CCR5-modified hiPSCs are shown in [Fig. 4(i)], while those of the biallele CCR5-modified hiPSCs are shown in Fig. 4(ii). The PCR fragments were then cloned and sequenced to determine the nucleotide sequence of the CCR5 mutations both in the single-allele CCR5-modified hiPSCs and biallele CCR5-modified hiPSCs, which are shown in Table 1. As expected, the Cre-treated single-allele CCR5-modified hiPSCs had one CCR5 wild-type allele and the other CCR5 allele was disrupted by a loxP site, while the Cre-treated biallele CCR5-modified hiPSCs had one CCR5 allele disrupted by an NHEJ mutation as before the Cre treatment and the other CCR5 allele was disrupted by a loxP site (Fig. 4; Table 1). Insertion of a loxP site within the CCR5 allele results in biallele CCR5-modified hiPSCs, with functional deletion of CCR5. In order to evaluate the pluripotency of hiPSCs, both before and after Cre-recombinase-mediated excision of the reprogramming cassette, quantitative reverse transcription (RT)-polymerase chain reaction was performed to compare the mRNA expression profiles before and after the removal of the transgenes (Fig. 5). The hiPSCs after removal of the stem cell factor transgenes continued to express the pluripotency genes from the endogenous chromosomal loci. As expected, IMR90 cells, which were used as a control, did not show expression of any pluripotency genes from the genomic loci.
FIG. 4.

PCR and nucleotide sequence analysis of the mutated CCR5 locus of single-allele and biallele CCR5-modified hiPSCs. (i) Schematic diagram showing the configuration of the CCR5 chromosomal locus in single-allele CCR5-modified hiPSCs before and after Cre treatment. PCR amplification of the mutant CCR5 locus was performed using genomic DNA from single-allele CCR5-modified hiPSCs and primers flanking the CCR5-specific ZFN target sites. PCR analysis yields the expected 900 bp fragment for the CCR5 allele with a loxP site insertion and the expected 854 bp fragment for the CCR5 wild-type (WT) allele. (ii) Schematic diagram showing the configuration of the CCR5 locus in the biallele CCR5-modified hiPSCs before and after Cre treatment. PCR amplification of the mutant CCR5 loci was performed using genomic DNA from the 4 biallele CCR5-modified hiPSCs and primers flanking the mutation site(s). PCR analysis yields the expected 900 bp fragment for the CCR5 allele with a loxP site insertion and the expected 849, 856, 846 and 851 fragments, respectively, for the CCR5 allele mutated by non homologous end joining (lanes 1–4) for the CCR5-CRE-B-hiPSC1, CCR5-CRE-B-hiPSC2, CCR5-CRE-B-hiPSC3 and CCR5-CRE-B-hiPSC4 lines. The PCR fragments were then cloned and sequenced to determine the nucleotide sequence at the mutated CCR5 locus (see Table 1). Color images available online at www.liebertpub.com/scd
Table 1.
Sequence Analysis of the CCR5 Locus in Single-allele and Biallele CCR5-Modified IMR90 hiPSCs, Post-CRE Treatment
 |
FIG. 5.

Quantitative reverse transcription (RT)-polymerase chain reaction analysis showing the expression levels of transgenes and endogenous genes IMR90 hiPSC lines. (i) Oct4; (ii) Sox2; (iii) Nanog; and (iv) Rex1 in single-allele biallele CCR5-modified hiPSCs, respectively, before and after the removal of reprogramming transcription factor genes cassette using Cre-recombinase. Expression levels of individual transcripts (Oct4, Sox2, Nanog and Rex1) were normalized to glyceraldehyde 3-phosphate dehydrogenase expression. IMR90 cells, which were used as a control, did not reveal expression of any pluripotency genes from their genomic loci. Fib denotes IMR90 cells; S-hiPSCs denotes single-allele CCR5-modified hiPSC lines; and B-hiPSCs denotes biallele CCR5-modified hiPSC lines.
Genetic engineering of the single-allele CCR5-modified hiPSCs by targeted insertion and expression of the large CFTR transcription unit from the remaining wild-type CCR5 allele, using CCR5-specific ZFNs
Nucleofection of single-allele CCR5-modified hiPSCs using tdTomato/CFTR cDNA donor and designed CCR-specific ZFNs resulted in the targeted addition of the CFTR transcription unit at the remaining wild-type CCR5 allele [Figs. 1(vi) and 6]. The hiPSCs expressing tdTomato were FACS sorted and single-cell colonies were isolated by serial dilution and grown [Fig. 6(i); Supplementary Fig. S2b]. The donor construct contained tdTomato gene under the control of the Pcmv promoter, while CFTR cDNA under the control of Pcag promoter [Fig. 6(ii)]. As expected, all the pluripotency markers were expressed in the single-cell colonies of tdTomato/CFTR hiPSCs that were examined [Fig. 6(i)]. PCR analysis of the 5′ and 3′ junctions of the donor insertion site were performed using appropriate primers (one of the primers anchored outside the CCR5 homology arms and the other anchored within the donor) as shown in Fig. 6(ii), which yielded the expected DNA fragments: 1.8 kb [Fig. 6(iii)] and 1.4 kb [Fig. 6(iv)], respectively, confirming tdTomato/CFTR donor insertion at the wild-type CCR5 allele. The PCR fragments were then cloned and sequenced to determine the nucleotide sequence at the 5′ and 3′ junctions of the donor insertion site, which revealed that tdTomato/CFTR donor was indeed inserted at the remaining wild-type CCR5 allele of the single-allele CCR5-modified hiPSCs (Table 2). Western blot profiles show efficient expression of the fully glycosylated CFTR from the endogenous CCR5 locus in all the tdTomato/CFTR hiPSCs that we examined as seen from the prominent C-band in Fig. 6(vi). Since the CFTR-expressing hiPSC lines were derived from single-cell colonies and grown over 15 passages, it appears that the CFTR expression is quite stable in these CCR5-modified hiPSC lines.
FIG. 6.

Targeted addition and expression of the large CFTR transcription unit from the wild-type CCR5 allele of the single-allele CCR5-modified IMR90 hiPSCs, using CCR5-specific ZFNs. (i) Morphology of single-allele CCR5-modified hiPSCs with targeted addition of the CFTR transcription unit at the remaining wild-type CCR5 allele. (A) Bright field images of the morphology of 3 single-cell CFTR hiPSC colonies. (B) tdTomato fluorescence images of CFTR hiPSC colonies shown in (A). Immunostaining for Oct4/Sox2/Nanog and DAPI staining of 3 representative CFTR hiPSCs are also shown. (ii) Schematic representation of donor (tdTomato/CFTR flanked by CCR5 homology arms) insertion site at the CCR5 locus of single-allele CCR5-modified hiPSCs. PCR primers anchored outside the CCR5 homology arms and primers anchored inside the donor for both 5′ and 3′ junction sites are shown. pA denotes polyA sequence. (iii) PCR analysis of 5′ junction of donor insertion site in 4 different CCR5-modified CFTR hiPSC single-cell colonies. Lanes: 1–5, CCR5-modified CFTR hiPSC colonies; 5, 1 kb ladder. PCR analysis yielded the expected band size (1.8 kb) confirming insertion of the donor at the remaining CCR5 wild-type allele. (iv) PCR analysis of 3′ junction of donor insertion site in 5 different CCR5-modified CFTR hiPSC single-cell colonies. Lanes: 1–5, CCR5-modified CFTR hiPSC colonies; 5, 1 kb ladder. PCR analysis yielded the expected band size (1.4 kb) confirming insertion of the donor at the remaining CCR5 WT allele in single-allele CCR5-modified hiPSCs. (v) The Southern blot profile of 4 different CCR5-modified CFTR-expressing hiPSCs, probed using labeled tdTomato gene. Lanes: 1, IMR90 cells; 2, control single-allele CCR5-modified hiPSCs before tdTomato/CFTR insertion at the remaining CCR5 WT allele; 3–6, 4 different CCR5-modified tdTomato/CFTR hiPSC lines; 7 and 8, lanes intentionally left blank; and 9, labeled tdTomato probe used as a positive control. All 4 CFTR-expressing hiPSCs show single integration of the CFTR transcription unit at the remaining WT CCR5 allele, which is consistent with the results obtained by PCR amplification using the corresponding locus-specific primers and sequencing of the 5′ and 3′ junctions of the donor insertion sites (Table 2). (vi) Western blot profile of 4 different CCR5-modified tdTomato/CFTR hiPSCs, probed using the antibody Ab217, anti-C-terminal monoclonal mouse antibody against CFTR (purchased from UNC Center for Cystic Fibrosis Research) is shown. Lanes: 1, IMR90 cells; 2, control single-allele CCR5-modified hiPSCs before tdTomato/CFTR insertion at the remaining CCR5 wild-type allele; and 3–6, 4 different single-cell colonies of CCR5-modified CFTR hiPSCs. Color images available online at www.liebertpub.com/scd
Table 2.
Sequence Analysis of 5′ and 3′ Junctions of the tdTomato/CFTR Donor Insertion Site in tdTomato/CFTR hiPSCs
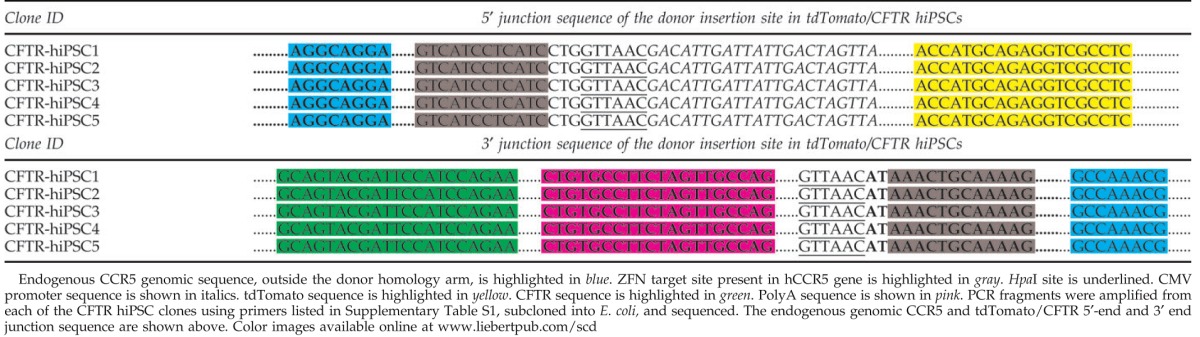 |
Teratoma formation and karyotyping of tdTomato/CFTR-expressing hiPSCs
CFTR-expressing hiPSCs were analyzed for chromosomal abnormality by karyotyping and for pluripotency by teratoma formation in nude mice, respectively (Fig. 7). Karyotyping of CFTR-expressing hiPSCs showed that the cells were normal, 46XX [Fig. 7(i)]. As expected, teratomas showed formation of all 3 germ layers [Fig. 7(ii)]. CFTR-expressing hiPSC lines maintained the expression of pluripotency markers for more than 15 passages and their ability to differentiate in vivo into derivatives of all 3 germ layers (ectoderm, mesoderm and endoderm), demonstrating that the genome modification did not alter the pluripotency of the CFTR-expressing hiPSCs.
FIG. 7.

Characterization of tdTomato/CFTR-expressing CCR5-modified hiPSCs. (i) Karyotyping of tdTomato/CFTR-expressing hiPSCs show that the cells are normal, 46XX. (ii) Teratoma formation shows that the tdTomato/CFTR-expressing hiPSCs produce all 3 germ layers, ectoderm, mesoderm and endoderm. Color images available online at www.liebertpub.com/scd
Off-target cleavage by CCR5-specific ZFNs
We also monitored the off-target effects of the CCR5-specific ZFNs fused to FokI nuclease domain variants (REL_DKK) in CCR5-modified tdTomato/CFTR-expressing hiPSCs at the closely related CCR2 locus and at 4 other previously reported CCR5 ZFN susceptible sites (KRR1, ALBIM2, PKN2, and PGC) by PCR amplification and followed by sequencing. The results are summarized in Supplementary Fig. S3 and Supplementary Table S5. No mutation was induced by CCR5-specific ZFNs at any of these chromosomal loci.
Generation of hiPSCs from human primary CBMNCs using CCR5-specific ZFNs
We generated both single-allele CCR5-modified hiPSCs and biallele CCR5-modified hiPSCs by targeted insertion of reprogramming genes (Oct4/Sox2/Klf4/Lin28/Nanog/eGFP) flanked by loxP sites, at the CCR5 locus of CBMNCs, using ZFN-mediated gene targeting (Fig. 8). Twenty individual single-cell colonies with morphology similar to hES cells were picked, grown, and characterized further for genotype of the CCR5 locus by PCR and sequencing. Complete data for 5 single-allele and biallele CCR5-modified CBMNC hiPSCs are shown in Supplementary Fig. S4 and Fig. 8, respectively. The CCR5-gene-modified hiPSC colonies were characterized for expression of pluripotency markers [Fig. 8(i); Supplementary Fig. S4(i)]. As expected, all the pluripotency markers were expressed in all the single-cell colonies that were examined. PCR analysis of the 5′ and 3′ junctions of the donor insertion site [using one of the primers anchored outside the CCR5 homology arms of the donor and the other anchored within the donor as shown in Supplementary Fig. S4(ii) for single-allele CCR5-modified hiPSCs; and as shown in Fig. 8(ii) for biallele CCR5-modified hiPSCs] yielded the expected 1.2 and 1.5 kb fragments respectively [Fig. 8(iii, iv); Supplementary Fig. S4(iii, iv)] indicating donor insertion at the CCR5 locus. The PCR fragments were then cloned and sequenced to determine the nucleotide sequence at the 5′ and 3′ junctions of the donor insertion site in the CCR5-modified hiPSCs. The sequencing data further confirmed the presence of both single-allele CCR5-modified hiPSCs resulting from HR (Supplementary Fig. S4; Supplementary Table S2b) and biallele CCR5-modified hiPSCs resulting from HR and NHEJ, (Fig. 8; Supplementary Fig. S1b; Supplementary Tables S2b and S3). The ratio of single-allele (15 single-cell colonies) versus biallele (5 single-cell colonies) CCR5-modified colonies for CBMNC-derived hiPSC lines was 3:1 (Supplementary Table S4b).
FIG. 8.

Characterization of biallele CCR5-modified hiPSCs, generated from cord blood mononuclear cells (CBMNCs) by targeted insertion of Oct4/Sox2/Klf4/Nanog/Lin28 transcription factors and eGFP at the CCR5 locus, using ZFNs. (i) Morphology of precisely targeted biallele CCR5-modified hiPSCs before CRE treatment. (A) Bright field images of the morphology 2 representative biallele CCR5-modified hiPSC colonies. (B) GFP fluorescence images of hiPSC single-cell colonies shown in (A). Immunostaining for Oct4/Sox2/Nanog/Tra1-60 and DAPI staining of the hiPSC lines are also shown. (ii) Schematic representation of donor (Oct4/Sox2/Klf4/Lin28/Nanog and eGFP flanked by 750 bp CCR5 homology arms) insertion site at the CCR5 locus of biallele CCR5-modified CBMNC hiPSCs. PCR primers anchored outside the CCR5 homology arms and primers anchored inside the donor for the 5′ and 3′ junction sites are shown. (iii) PCR analysis of 5′ junction of donor insertion site in 5 different biallele CCR5-modified hiPSC lines. Lanes: 1, control CBMNCs; 2–6, biallele CCR5-modified hiPSC lines; and 7, 1 kb ladder. PCR analysis yields the expected band size (1.2 kb) confirming insertion of the donor at the CCR5 locus. (iv) PCR analysis of 3′ junction of donor insertion site in 5 different single-allele CCR5-modified hiPSC single-cell colonies. Lanes: 1, Control CBMNCs; 2–6, biallele CCR5-modified hiPSC colonies; and 7, 1 kb ladder. PCR analysis yields the expected band size (1.5 kb) confirming insertion of the donor at the CCR5 locus. (v) Southern blot profiles of 5 different single-allele and biallele CCR5-modified hiPSC lines, respectively. Genomic DNA was digested with BamHI and hybridized using either 32P-radiolabeled eGFP gene or 32P-radiolabeled Sox2 gene. Lanes: 1, Control CBMNCs; 2–6, 5 different single-allele CCR5-modified hiPSC lines; 7–11, 5 different biallele CCR5-modified hiPSC lines. Immunostaining and PCR analysis of single-allele CCR5-modified CBMNC hiPSC lines are shown in Supplementary Fig. S4. Target denotes donor (containing Sox2 gene) insertion at the CCR5 locus; Wt denotes endogenous Sox2 genes. Color images available online at www.liebertpub.com/scd
Monitoring off-target integrations of CCR5-modified hiPSCs (generated from IMR90 cells and CBMNCs using ZFNs) and tdTomato/CFTR-expressing hiPSCs by Southern
Southern blot analysis of genomic DNA (isolated from single-allele and biallele CCR5-modified IMR90 hiPSCs) digested with BamHI and probed using either 32P-radiolabeled eGFP gene or 32P-radiolabeled Sox2 gene confirmed single insertion of the donor at the targeted CCR5 locus [Fig. 2(vi)]. Southern blot analysis of CFTR-expressing hiPSCs using labeled tdTomato probe revealed only a single band, suggesting a single targeted integration event and ruling out random integration events within the genome of these tdTomato/CFTR-expressing hiPSC lines [Fig. 6(v)]. This result, taken together with the PCR and sequencing analysis of the donor integration site discussed above, confirms targeted integration of tdTomato/CFTR transcription unit at the CCR5 locus of CFTR hiPSCs. Southern blot analysis of genomic DNA (isolated from single-allele and biallele CCR5-modified CBMNC hiPSCs) digested with BamHI and probed using either 32P-radiolabeled GFP gene or 32P-radiolabeled Sox2 gene confirmed single insertion of the donor at the targeted CCR5 locus [Fig. 8(v)].
Discussion
In this article, we have shown that (1) ZFN-mediated gene targeting could be used to generate genetically well-defined hiPSCs from human lung fibroblasts and CBMNCs by targeted insertion of pluripotency genes into one of the CCR5 alleles; and (2) the functionality of such reprogrammed single-allele CCR5-modified hiPSCs (after the removal of the pluripotency genes by Cre treatment) could be further genetically engineered by targeted insertion of a potential therapeutic gene at the remaining wild-type CCR5 allele.
Generation of hiPSCs from human lung fibroblasts and CBMNCs using designed CCR5-specific ZFNs
While the development of integration-free reprogramming technology may offer a better approach to generate hiPSCs, here we investigated reprogramming of human lung fibroblasts by targeted addition of stem cell factor genes to a specific chromosomal locus using CCR5-specific ZFNs. Generation of iPSCs from human and murine cells using other similar methods such as piggybac transposons, phage integrase (ΦC31), and excisable lentiviruses have been reported elsewhere [32,33]. Although the ΦC31-derived hiPSCs had only a single integration in each line, the locations of integration were random in different lines, favoring intergenic regions [32]. It is quite possible that site-specific integration using ΦC31 integrase could result in insertions at critical genes or control regions in targeted cells disrupting normal function of these cells. This is also likely to be true of excisable lentiviral approach, since “scarred” genomic sequences will be left behind at the site of integration after lentiviral excision, which could disrupt normal function of the cells. In our experiments using CCR5-specific ZFNs, we observed integration of the pluripotency genes only at the CCR5 locus in all the 4 single-colony hiPSCs that we examined. Thus, unlike the ΦC31 integrase-mediated approach, ZFN-mediated gene targeting resulted in highly specific integration of the pluripotency genes into the targeted CCR5 locus of the human lung fibroblasts, although at low efficiency (Supplementary Table S4). Further improvements to reprogramming efficiency could be achieved by reducing the number of plasmids used in co-transfection to 2 (by cloning both ZFNs in a single plasmid plus the donor plasmid) from 3 (2 ZFN plasmids plus the donor plasmid). More recently, we have also achieved reprogramming of human primary CBMNCs (with >50% CD34+ cells, purchased from AllCells, Inc.) by a single co-transfection with CCR5-specific ZFNs and a donor containing 5 stem cell factor genes. The reprogramming efficiency of human primary CBMNCs using 5 stem cell factor genes (Oct4/Sox2/Klf4/Lin28/Nanog) was similar to that of IMR90 cells. The observed frequency of biallele CCR5-modified hiPSCs was about 20%–25% using these cells.
Biallele CCR5-modified mutant hiPSCs
ZFN-mediated gene targeting of the CCR5 locus using a donor encoding reprogramming factors yielded both single-allele CCR5-modified hiPSCs (with a single-allele insertion of the pluripotency genes at the CCR5 locus, while the second CCR5 allele remained wild type) and biallele CCR5-modified mutant hiPSCs (with a single-allele insertion of the pluripotency genes at the CCR5 locus, while the second CCR5 allele was mutated by NHEJ). Since Cre treatment of the latter results in the disruption of both CCR5 alleles leading to functional deletion of CCR5, in theory, such biallele CCR5-modified hiPSC colonies could be isolated and differentiated into HIV-resistant CD34+ hematopoietic stem cells (HSCs) for transplantation. However, technical challenges still remain for the differentiation of hiPSCs into HSCs [34]. The success of this approach depends on being able to get hiPSCs to make in vivo engrafting HSCs for hematopoietic reconstitution, which has not yet been achieved [34]. However, once these challenges are overcome, we posit that ZFN-mediated disruption of CCR5 locus by HR in patient-specific hiPSCs, would become the method of choice for generation of HIV-resistant CD34+ cells for autologous transplantation via differentiation of HIV-resistant biallele CCR5-modified hiPSC colonies generated either from cord blood or from human fibroblasts. Thus, ZFN-mediated approach has the potential to generate hiPSC clones containing uniform biallele CCR5-modified cells containing a specific mutation. HR-driven disruption of CCR5 using targeted insertion of the transgenes like puromycin at the CCR5 locus could also be used to generate and select a specific mutant population of biallele CCR5-modified hiPSCs.
Holt et al. have reported high efficiency ZFN-mediated disruption of the CCR5 locus by NHEJ in human CD34+ HSPCs harvested from umbilical cord blood (17% of the total CCR5 alleles in a population of cells was shown to contain both single-allele and biallele CCR5-modified cells) [23–25]. Holt et al. then showed that ZFN treated HSPCs retained the ability to engraft in NSG (NOD/SCID/IL2rγnull) mice and gave rise to polyclonal multi-lineage progeny cells in which CCR5 was permanently disrupted [25]. Furthermore, in mice transplanted with ZFN-modified HSPCs, there was rapid selection for CCR5−/− cells and these mice had significantly lower HIV-1 levels [25]. However, it must be emphasized that ZFN-mediated CCR5 disruption of CD34+ cells by NHEJ produces a heterogeneous population of cells containing a spectrum of CCR5 mutations, of which only a small percentage are likely to result in the disruption of CCR5. Even a much smaller percentage of the CCR5-gene-modified cells would have biallele disruption of CCR5 that confers resistance to HIV-1 infection. Therefore, we reason that the single-cell hiPSC colony-derived homogeneous CD34+ cell population with a defined biallele CCR5-mutation is likely to more advantageous in transplantation studies as compared to a heterogeneous CD34+ cell population generated by NHEJ.
Targeted CFTR gene addition to the CCR5 safe harbor locus of the human genome
Genetic engineering of hiPSCs using designed ZFNs or TALENs have been reported previously in literature employing hiPSCs generated by random integrations of pluripotency genes [21–25] using retroviral methods, rather than site-specific integration. ZFN-mediated approach, however, generates precisely targeted genetically well-defined hiPSCs, circumventing the problems associated with random integrations. Targeting of selectable marker transgenes (like GFP or puromycin) to the IL2RG, PPP1R12C, AAVS1, and CCR5 loci have been described previously elsewhere [21,24,25].
Since our ultimate goal includes autologous cell-based transgene-correction therapy for recessive monogenic human diseases, targeting a therapeutic gene to a safe and specified genomic location offers a more general approach to treat a variety of monogenic diseases unlike the patient-specific gene correction approach. Such an approach will also ensure that the therapeutic gene will not be expressed from an endogenous promoter, circumventing the risk of insertional mutagenesis that could result from random uncontrolled integration events. Therefore, the successful experiment on site-specific addition and expression of the large CFTR transcription unit (a potential therapeutic gene) from the remaining wild-type CCR5 allele of the single-allele CCR5-modified hiPSCs is very encouraging. The facts that integration of the therapeutic gene to the CCR5 locus was efficient, that tdTomato gene expression can readily be used as a marker to sort out the CCR5-gene-modified hiPSCs by FACS, and that high expression of CFTR was observed in all the 4 single-cell colony hiPSCs that we examined confirm that the CCR5 locus could serve as an ideal locus for targeted addition and ectopic expression to achieve therapeutic transgene correction by functional protein complementation in cells with corresponding gene defects, for example, in cells with sickle cell disease or cystic fibrosis (CF) disease. However, how high expression of the therapeutic protein will affect pluripotency and differentiation propensity of hiPSCs with the corresponding gene defect is unknown at this time and needs to be studied in detail in the future. The use of embryonic and adult stem cells for lung repair and regeneration after injury has been proposed as a potential therapeutic approach for CF and other lung diseases [35]. However, there still remains the technical challenge of delivering the transgene-corrected cells to the lung that needs to be addressed before any cell-based therapy for CF treatment could even be contemplated [36]. This study at this juncture is a demonstration of the potential of the ZFN technology to generate and further reshape the functionality of hiPSCs. As an alternate approach, CFTR-specific ZFNs that bind and cleave near the Δ508F mutation in the CFTR gene have been generated in the lab for potential use as agents for in situ correction of CFTR defects in lung cells. During the revision of this article, Ciaran et al. reported correction of the ΔF508 mutation in the CFTR gene by ZFN homology-directed repair in human cells expressing CFTR [37].
A potential limitation of the ZFN-mediated targeting approach is off-target DNA cleavage at related sequences (like the CCR2 locus when targeting CCR5), which may cause unpredictable genotoxic effects [4,38,39]. In this study, we used only the well-characterized highly specific 4-finger CCR5-specific ZFNs, which were previously shown not to result in additional integrations [4,22,23]. Southern blot analysis of the CCR5-modified CFTR-expressing hiPSCs also did not reveal any additional integration events. Sequencing of the CCR2 site closely related to the CCR5 and 4 other previously reported susceptible sites (KRR1, ABLIM2, PKN2, and PGC) [38] of the CCR5-modified CFTR-expressing hiPSCs also did not reveal any mutations at these loci (Supplementary Fig. S2; Supplementary Table S5). Karyotyping results of CCR5-modified CFTR-expressing hiPSCs also confirmed that these cells are normal 46XX, ruling out any chromosomal abnormality. However, ZFN-mediated DSB (or TALEN-mediated DSB) and error-prone repair by NHEJ elsewhere in the genome still needs to be examined in great detail on a genome-wide basis, especially if human therapeutics studies are being contemplated using ZFN-mediated (or TALEN-mediated) approach [38–41]. This may necessitate very detailed genome-wide analysis of multiple patient-specific single-cell hiPSC colonies before the correct one could be identified.
In ex vivo gene therapy, autologous stem cells from a patient with a genetic disease are isolated, gene corrected, expanded in vitro, and then reimplanted into the patient. The limited availability of patient-derived adult stem cells and their limited in vitro expansion potential have greatly hindered this method in clinical practice. ZFN-evoked strategies described here to generate precisely targeted genetically well-defined hiPSCs using designed ZFNs (or TALENs) and the ability to subsequently regulate and reshape the functionality of such hiPSCs by targeted insertion and high expression of therapeutic genes from the endogenous CCR5 safe-harbor locus of the human genome for functional protein complementation may have great potential for use in a clinical setting to treat a various recessive monogenic diseases. Furthermore, CCR5-modified hESCs and CCR5-modified hiPSCs were recently shown to retain their pluripotent characteristics and could be differentiated in vitro into CD34+ cells that formed all types of hematopoietic colonies [42]. This suggests the potential for using patient-specific biallele CCR5-modified hiPSC-derived CD34+ cells (generated by using ZFN-evoked strategies) to treat HIV/AIDS.
Supplementary Material
Acknowledgments
This work was supported by a grant from National Institute of General Medical Sciences (GM077291) and by a Phase I grant to K.K. from the Bill & Melinda Gates Foundation through the Grand Challenge Explorations Initiative. S.R. is supported by an MSCRF post-doctoral fellowship award and exploratory research grant. We thank Mr. S. Gunasegaran (President and CEO, PBPL, India) for his constant support and encouragement. We thank Dr. Robert Siliciano for encouragement and helpful suggestions with the CCR5 disruption experiments. We thank Drs. Fred Bunz, Elias Zambidis, and Linzhou Cheng for helpful suggestions with hiPSC culturing and immunostaining experiments and Dr. Margolick's lab for assistance with the flow cytometry and FACS studies.
Author Disclosure Statement
The authors declare no conflicts of interest.
References
- 1.Kim Y-G. Cha J. Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bibikova M. Carroll D. Segal DJ. Trautman JK. Smith J. Kim YG. Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage by a chimeric nuclease. Mol Cell Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sander JD. Dahlborg EJ. Goodwin MJ. Cade L. Zhang F. Cifuentes D. Curtin SJ. Blackburn JS. Thibodeau-Beganny S, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat Methods. 2011;8:67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramalingam S. Kandavelou K. Rajenderan R. Chandrasegaran S. Creating designed zinc finger nucleases with minimal cytotoxicity. J Mol Biol. 2011;405:630–641. doi: 10.1016/j.jmb.2010.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cermak T. Doyle EL. Christian M. Wang L. Zhang Y. Schmidt C. Baller JA. Somia NV. Bogdanove AJ. Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng X. Noyes MB. Zhu LJ. Lawson ND. Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26:695–701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porteus MH. Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 8.Urnov FD. Miller JC. Lee YL. Beausejour CM. Rock JM. Augustus S. Jamieson AC. Porteus MH. Gregory PD. Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 9.Kandavelou K. Mani M. Durai S. Chandrasegaran S. ‘Magic’ scissors for genome surgery. Nat Biotechnol. 2005;23:686–687. doi: 10.1038/nbt0605-686. [DOI] [PubMed] [Google Scholar]
- 10.Wu J. Kandavelou K. Chandrasegaran S. Custom-designed zinc finger nucleases: what is next? Cell Mol Life Sci. 2007;64:2933–2944. doi: 10.1007/s00018-007-7206-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moehle EA. Rock JM. Lee YL. Jouvenot Y. DeKelver RC. Gregory PD. Urnov FD. Holmes MC. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci U S A. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kandavelou K. Ramalingam S. London V. Mani M. Wu J. Alexeev V. Civin CI. Chandrasegaran S. Targeted manipulation of mammalian cells using designed zinc finger nucleases. Biochem Biophys Res Commun. 2010;388:56–61. doi: 10.1016/j.bbrc.2009.07.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connelly JP. Barker JC. Pruett-Miller S. Porteus MH. Gene correction by homologous recombination with zinc finger nucleases in primary cells from a mouse model of a generic recessive genetic disease. Mol Ther. 2010;18:1103–1110. doi: 10.1038/mt.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou J. Maeder ML. Mali P. Pruett-Miller SM. Thibodeau-Beganny S. Chou BK. Chen G. Ye Z. Park IH, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;2:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabastiano V. Maeder ML. Angstman JF. Haddad B. Khayter C. Yeo DT. Goodwin MJ. Hawkins JS. Ramirez CL, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29:1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yusa K. Rashid ST. Strick-Marchand H. Varela I. Liu PQ. Paschon DE. Miranda E. Ordóñez A. Hannan NR, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hockemeyer D. Soldner F. Beard C. Gao Q. Mitalipova M. DeKelver RC. Katibah GE. Amora R. Boydston EA, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hockemeyer D. Wang H. Kiani S. Lai CS. Gao Q. Cassady JP. Cost GJ. Zhang L. Santiago Y, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou J. Sweeney CL. Chou BK. Choi U. Pan J. Wang H. Dowey SN. Cheng L. Malech HL. Oxidase deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease mediated safe harbor targeting. Blood. 2011;117:5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeKelver RC. Choi VM. Moehle EA. Paschon DE. Hockemeyer D. Meijsing SH. Sancak Y. Cui X. Steine EJ, et al. Functional genomics, proteomics, regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20:1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bobis-wosowicz S. Osiak A. Rahman SH. Cathomen T. Targeted genome editing in pluripotent stem cells using zinc-finger nucleases. Methods. 2011;53:339–346. doi: 10.1016/j.ymeth.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 22.Lombardo A. Genovese P. Beausejour CM. Colleoni S. Lee YL. Kim KA. Ando D. Urnov FD. Galli C, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 23.Perez EE. Wang J. Miller JC. Jouvenot Y. Kim KA. Liu O. Wang N. Lee G. Bartsevich VV, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilen CB. Wang J. Tilton JC. Miller JC. Kim KA. Rebar EJ. Sherrill-Mix SA. Patro SC. Secreto AJ, et al. Engineering HIV-resistant human CD4+ T Cells with CXCR4-specific zinc-finger nucleases. PLoS Pathogens. 2011;7:e1002020. doi: 10.1371/journal.ppat.1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holt N. Wang J. Kim K. Friedman G. Wang X. Taupin V. Crooks GM. Kohn DB. Gregory PD. Holmes MC. Cannon PM. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller JC. Holmes MC. Wang J. Guschin DY. Lee YL, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 27.Szczepek M. Brondani V. Buchel J. Serrano L. Segal DJ. Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 28.Carey BW. Markoulaki S. Hanna J. Saha K. Gao Q. Mitalipova M. Jaenisch Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci U S A. 2009;106:157–162. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibson DG. Young L. Chuang RY. Venter JC. Hutchison CA., 3rd Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 30.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 31.Chou BK. Mali P. Huang X. Ye Z. Dowey SN. Resar LM. Zou C. Zhang YA. Tong J. Cheng L. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21:518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye L. Chang JC. Lin C. Qi Z. Yu J. Kan YW. Generation of induced pluripotent stem cells using site-specific integration with phage integrase. Proc Natl Acad Sci U S A. 2010;107:19467–19472. doi: 10.1073/pnas.1012677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karow M. Chavez CL. Farruggio AP. Geisinger JM. Keravala A. Jung WE. Lan F. Wu JC. Chen-Tsai Y. Calos MP. Site-specific recombinase strategy to create induced pluripotent stem cells efficiently with plasmid DNA. Stem Cells. 2011;29:1696–1704. doi: 10.1002/stem.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKinney-Freeman SL. Daley GQ. Towards hematopoietic reconstitution from embryonic stem cells: a sanguine future. Curr Opin Hematol. 2007;14:343–347. doi: 10.1097/MOH.0b013e3281900edd. [DOI] [PubMed] [Google Scholar]
- 35.Weiss DJ. Stem cells and cell therapies for cystic fibrosis and other lung diseases. Pulm Pharmacol Ther. 2008;21:588–594. doi: 10.1016/j.pupt.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakland M. Sinn PL. McCray PB., Jr. Advances in cell and gene-based therapies for cystic fibrosis lung disease. Mol Ther. 2012 doi: 10.1038/mt.2012.32. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciaran ML. Rowan F. Jennifer AH. Martina FS. Patrick TH. Correction of the ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. BioResearch. 2012;3:99–108. doi: 10.1089/biores.2012.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabriel R. Lombardo A. Arens A. Miller JC. Genovese P. Kaeppel C. Nowrouzi A. Bartholomae CC. Wang J, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 39.Pattanayak V. Ramirez CL. Joung JK. Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeder ML. Thibodeau-Beganny S. Osiak A. Wright DA. Anthony RM. Eichtinger M. Jiang T. Foley JE. Winfrey RJ, et al. Rapid “open-source” engineering of customized zinc finger nucleases for highly efficient modification. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reyon D. Tsai SQ. Khayter C. Foden JA. Sander JD. Joung KJ. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012 doi: 10.1038/nbt.2170. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao Y. Nashun B. Zhou T. Qin L. Qin L. Zhao S. Xu J. Esteban MA. Chen X. Generation of CD34(+) cells from CCR5-disrupted human embryonic and induced pluripotent stem cells. Hum Gene Ther. 2011;23:238–242. doi: 10.1089/hum.2011.126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.