

The crystal structure of quercitrin, a naturally occurring flavonol glycoside, has been determined in a complex with the N-terminal kinase domain of murine RSK2. The structure revealed that quercitrin inhibits the RSK2 kinase in the same fashion as another known inhibitor, SL0101.
Keywords: protein kinases, inhibitors, flavonol glycosides, quercitrin, SL0101
Abstract
Members of the RSK family of kinases constitute attractive targets for drug design, but a lack of structural information regarding the mechanism of selective inhibitors impedes progress in this field. The crystal structure of the N-terminal kinase domain (residues 45–346) of mouse RSK2, or RSK2NTKD, has recently been described in complex with one of only two known selective inhibitors, a rare naturally occurring flavonol glycoside, kaempferol 3-O-(3′′,4′′-di-O-acetyl-α-l-rhamnopyranoside), known as SL0101. Based on this structure, it was hypothesized that quercitrin (quercetin 3-O-α-l-rhamnopyranoside), a related but ubiquitous and inexpensive compound, might also act as an RSK inhibitor. Here, it is demonstrated that quercitrin binds to RSK2NTKD with a dissociation constant (K d) of 5.8 µM as determined by isothermal titration calorimetry, and a crystal structure of the binary complex at 1.8 Å resolution is reported. The crystal structure reveals a very similar mode of binding to that recently reported for SL0101. Closer inspection shows a number of small but significant differences that explain the slightly higher K d for quercitrin compared with SL0101. It is also shown that quercitrin can effectively substitute for SL0101 in a biological assay, in which it significantly suppresses the contractile force in rabbit pulmonary artery smooth muscle in response to Ca2+.
1. Introduction
Naturally occurring flavonoids, all of which share a common core consisting of an aromatic ring, a benzopyran ring and a phenyl substituent, are secondary metabolites that provide UV protection and color to plants. They are found in virtually all fruits and vegetables (Hou & Kumamoto, 2010 ▶). The most ubiquitous among the flavonoids are flavonols, which are derivatives of 3-hydroxy-2-phenylchromen-4-one, and include, amongst others, kaempferol (3,4′,5,7-tetrahydroxy-2-phenylchromen-4-one), quercetin (3,3′,4′,5,7-pentahydroxy-2-phenylchromen-4-one), myricetin (3,3′,4′,5′,5,7-hexahydroxy-2-phenylchromen-4-one) and fisetin (3,3′,4′,7-tetrahydroxy-2-phenylchromen-4-one). Flavonols are under intense scrutiny as key factors in the reduced risk of cancer and cardiovascular disease that is associated with plant-rich diets (Calderón-Montaño et al., 2011 ▶; Neuhouser, 2004 ▶; Maron, 2004 ▶).
Known primarily for their antioxidant properties, flavonols are also increasingly recognized as inhibitors of kinases, albeit with relatively low affinity and selectivity (Hou & Kumamoto, 2010 ▶; Williams et al., 2004 ▶). For example, kaempferol inhibits myosin light-chain kinase (Rogers & Williams, 1989 ▶) and phosphatidylinositol 3-kinase (Lee et al., 2010 ▶). Quercetin has been found to inhibit 16 kinases relevant to cancer-cell growth (Boly et al., 2011 ▶). Myricetin inhibits Akt (Kim et al., 2010 ▶) and phosphoinositide 3-kinase (Walker et al., 2000 ▶), while fisetin inhibits Akt and rapamycin kinase (mTOR; Suh et al., 2010 ▶). Several crystal structures of complexes of protein kinase domains with flavonols are known, including those of Pim1 kinase with quercetin, myricetin and quercetagetin (Holder et al., 2007 ▶), phosphoinositide 3-kinase with quercetin and myricetin (Walker et al., 2000 ▶) and HCK kinase with quercetin (Sicheri et al., 1997 ▶). These crystallographic studies show that flavonols are type I kinase inhibitors, i.e. they bind within the ATP-binding pocket so that the flavonol mimics the binding mode of the adenine moiety of ATP.
The vast majority of flavonols are synthesized in plants as glycosides, with the 3- and 7-hydroxyls serving as glycosylation sites. Flavonol glycosides have not been studied as intensely as their aglycone counterparts with respect to their ability to interact with and inhibit kinases, but it is known that at least some of them have such properties. For example, luteolin-7-O-glucoside has been shown to be a highly selective inhibitor of JNK3 kinase (Goettert et al., 2010 ▶).
Recently, a new flavonol glycoside, kaempferol 3-O-(3′′,4′′-di-O-acetyl-α-l-rhamnopyranoside), also called SL0101, has been identified and shown to inhibit members of the p90 ribosomal S6 kinase (RSK) family (Smith et al., 2005 ▶; Bain et al., 2007 ▶; Fig. 1 ▶). The RSK kinases, of which four isoforms (RSK1–4) are known, are unique in that they have two distinct kinase domains in tandem (Anjum & Blenis, 2008 ▶; Pearce et al., 2010 ▶; Jones et al., 1988 ▶). Except for RSK4, which is constitutively active, RSK kinases are activated in response to growth factors, cytokines and other stimuli. Briefly, the process is initiated by the ERK1/2 kinases through phosphorylation of the activation loop of the C-terminal kinase domain (Thr577; human RSK2 numbering throughout), which belongs to the Ca2+/calmodulin-dependent kinase family. The activated C-terminal kinase domain (CTKD) catalyzes cis-phosphorylation of Ser369, which is within the hydrophobic motif downstream of the N-terminal kinase domain (NTKD), which is a member of the AGC kinase family (Pearce et al., 2010 ▶). This creates a docking site for the PDK1 kinase, which phosphorylates Ser227 (RSK2 numbering) within the activation loop of NTKD. The activated NTKD exerts the biological function of RSK by phosphorylating a plethora of downstream targets mostly involved in gene regulation (Anjum & Blenis, 2008 ▶). The SL0101 inhibitor binds exclusively to the NTKD.
Figure 1.

The chemical structures of the inhibitor SL0101 and its analogue quercitrin. The conformational dihedral angles ω, ϕ and ψ are also shown, together with the canonical numbering scheme of the whole molecule.
RSK kinases have recently attracted considerable attention specifically because of their involvement in cancer (Stratford & Dunn, 2011 ▶; Romeo & Roux, 2011 ▶). RSK1 and RSK2 have been found in a number of tumors, including breast (Smith et al., 2005 ▶), prostate (Clark et al., 2005 ▶) and head and neck tumors (Kang et al., 2010 ▶), as well as in multiple myelomas (Cuadrado & Nebreda, 2007 ▶; Kang et al., 2007 ▶), T-cell lymphomas (Kang et al., 2009 ▶) and melanomas (Mirmohammadsadegh et al., 2007 ▶). Links to other diseases are also important. For example, RSK1 has been implicated in ischaemia/reperfusion-induced injury (Maekawa et al., 2006 ▶).
Given this biomedical context, there is intense interest in the design of specific inhibitors for the RSK family of kinases (Nguyen, 2008 ▶). However, structure-based inhibitor design based on the SL0101 template has been impeded by a lack of direct structural information. In its absence, it has been simply assumed that SL0101 binds to NTKD so that its kaempferol moiety is inserted into the ATP-binding site in the canonical fashion, mimicking the adenine, which leaves the carbohydrate group exposed to the solvent (Nguyen et al., 2006 ▶; Nguyen, 2008 ▶; Cho et al., 2009 ▶; Lu et al., 2011 ▶).
Recently, we reported a crystal structure of the complex of the N-terminal domain of mouse RSK2 kinase (mRSK2NTKD) with SL0101 and showed that although the inhibitor binds within the ATP-binding site, it does so in a highly unusual manner with concomitant large conformational rearrangements in the protein (Utepbergenov et al., 2012 ▶). Specifically, the entire N-lobe, the hinge region between the lobes and the αD helix undergo dramatic conformational changes that together result in rearrangement of the nucleotide-binding site and the formation of a unique and highly hydrophobic pocket that accommodates SL0101.
SL0101 was initially isolated from extracts of the rare plant Forsteronia refracta found in the Amazon forest, but has subsequently been synthesized chemically (Maloney & Hecht, 2005 ▶). It is a useful but expensive reagent in studies of the cell biology of RSK kinases (approximately $250 per milligram). While enzymatic assays show that acylation of the rhamnose enhances the inhibitory potency of SL0101, the crystal structure of the complex revealed that the acetyl groups point into the solvent and make no significant contacts with the protein. We also reported the structure of the deacylated variant of SL0101, known as afzelin, and found that it binds to mRSK2NTKD in an identical way to SL0101. This result prompted speculation that another natural flavonol rhamnoside, quercitrin, may also function as an RSK inhibitor. Quercitrin differs from afzelin in that it has an additional hydroxyl at the 3′ position (Fig. 1 ▶). Unlike SL0101, quercitrin is a ubiquitous natural compound that is used as a dietary supplement and can be obtained commercially at much lower cost (∼$2.00 per milligram).
In this paper, we show that quercitrin binds to the isolated mRSK2NTKD, albeit with a slightly higher K d (dissociation constant) than SL0101. The binding mode of quercitrin, visualized here by a 1.8 Å resolution X-ray structure, is similar to that reported for SL0101. Although the differences are small, they are clearly defined by the electron density. We identify two specific close interatomic contacts between quercitrin and the protein which are probably responsible for the lower affinity of quercitrin towards mRSK2NTKD compared with SL0101. Furthermore, we discuss intriguing evidence that in their complexes with mRSK2NTKD both SL0101 and quercitrin are found in a high-energy conformation compared with the unbound free state, which is consistent with the notion of a complex induced-fit binding model that was first suggested in our paper describing the complex involving SL0101 (Utepbergenov et al., 2012 ▶).
Finally, we demonstrate that in spite of these small differences and the higher K d, quercitrin shows the same biological effects in a select functional assay (i.e. smooth-muscle contractility) as the synthetic SL0101 compound, suggesting that quercitrin may be used in cell biology as an inexpensive SL0101 surrogate.
2. Materials and methods
2.1. Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was performed at 298 K using a Microcal ITC-200 instrument (MicroCal, Northampton, Massachusetts, USA). The mRKS2NTKD samples were dialyzed against a buffer solution consisting of 50 mM Tris pH 8.0, 600 mM NaCl, 5 mM β-mercaptoethanol, 5% ethylene glycol prior to the experiment and all ligands were dissolved in the same buffer. The contents of the sample cell were stirred continuously at 700 rev min−1 during the experiment. A typical titration of mRSK2NTKD involved 18–22 injections of ligand (0.2–0.6 mM, 2 µl) into a sample cell containing 0.2 ml NTKDRSK2 (60–150 µM). The baseline-corrected data were analyzed with MicroCal Origin 5.0 software to determine the enthalpy change (ΔH), the association constant (K a) and the stoichiometry of binding (N) by fitting to the association model for a single set of identical sites. The protein was expressed and purified as described below, while quercitrin was purchased from Sigma–Aldrich.
2.2. Expression and purification of isolated mRSK2NTKD
The expression and purification of the N-terminal domain of murine RSK2, mRSK2NTKD (amino acids 45–346), has been described elsewhere (Utepbergenov et al., 2012 ▶). Briefly, the cDNA was cloned into pHisUni1 (Sheffield et al., 1999 ▶) vector using Phusion polymerase. The protein was overexpressed in Escherichia coli BL21 (RIPL) cells, isolated using His-Select nickel resin (Sigma–Aldrich), digested with rTEV protease and purified by nickel-affinity and size-exclusion chromatography. A cloning artifact, Gly-Ala-Met, was left at the N-terminus. The protein was mixed with quercitrin (40 mM solution in ethylene glycol) using an ∼10% excess of ligand, dialyzed against a buffer solution consisting of 50 mM Tris pH 8.0, 600 mM NaCl, 5 mM β-mercaptoethanol, 5 mM EDTA and used in crystallization setups. The final protein concentration was 5 mg ml−1.
2.3. Determination of the mRSK2NTKD–quercitrin crystal structure
Crystals of the mRSK2NTKD–quercitrin complex grew in 2–3 d at room temperature from vapour-diffusion setups prepared with the use of a Mosquito robot and consisting of equal volumes (250 nl) of the complex solution and a reservoir buffer consisting of 0.1 M HEPES pH 7.5, 30% Jeffamine ED-2003 (60 µl). Crystals were harvested in the reservoir buffer and flash-cooled in liquid nitrogen. Single-wavelength (λ = 1.000 Å) X-ray diffraction data were collected at 100 K on the SER-CAT (Southeast Regional Collaborative Access Team) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory, Chicago, USA. The data were indexed, integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶). R free was monitored by setting aside 5% of the reflections as a test set. Initial phase estimates were obtained by automated molecular replacement with BALBES (Long et al., 2008 ▶). A large part of the model was automatically built with ARP/wARP (Cohen et al., 2008 ▶) and was further improved manually with Coot (Emsley & Cowtan, 2004 ▶). Restrained positional and isotropic atomic displacement parameter (ADP) refinement was performed with PHENIX (Adams et al., 2010 ▶). CIF dictionaries for quercitrin were generated with eLBOW using the crystal structure of free quercitrin (Jiang et al., 2009 ▶). Figures were prepared using PyMOL (http://www.pymol.org/).
To identify the lowest energy conformation of quercitrin, a systematic search on its bound conformation in torsion space around the three dihedral angles (ω, ϕ and ψ; Fig. 1 ▶) was performed with SYBYL-X 1.3 (http://www.tripos.com). All possible conformers were generated for the three rotatable bonds at 10° angle increments starting at 0°. The generated conformers were subjected to energy-minimization in vacuum under the TRIPOS force field using Gasteiger–Hückel charges, a termination gradient set to 0.005 kcal mol−1 or a maximum of 10 000 iterations. The crystallographic conformation of free quercitrin was also subject to similar minimization steps. Also, using the RSK2 protein as an aggregate, the quercitrin structure was energy-minimized in situ in the protein pocket. The calculated strain energies are defined as the energy differences between the bound conformations of quercitrin and its global minimum.
2.4. Smooth-muscle contractility assays
All procedures were carried out according to protocols approved by the Animal Care and Use Committee at the University of Virginia. The primary branches of the rabbit pulmonary artery were cut into small strips (150–250 µm wide, 2–3 mm long) and mounted between two tungsten hooks on a bubble plate for force measurements (Horiuti, 1988 ▶; Kitazawa et al., 1989 ▶). The strips were stretched by 10% of their resting length and equilibrated for 40 min followed by permeabilization with 500 U ml−1 Staphylococcus aureus α-toxin prepared in our laboratory or 75 µM β-escin for 40 min at room temperature in relaxing solution (G1) consisting of 4.5 mM Mg–ATP, 1 mM EGTA as described previously (Kitazawa et al., 1989 ▶). To deplete Ca2+ stores, the strips were treated with 10 µM Ca2+ ionophore A23187 (Calbiochem) for 10–15 min in G1. For Ca2+-activating solutions, 10 mM EGTA was used and a calculated amount of calcium methanesulfonate was added to give the desired free Ca2+ concentration. Measurements of IC50 for RSK inhibitors were carried out in muscle strips stimulated with pCa 6.7. Once the force reached a plateau, increasing concentrations of inhibitor or the diluent (DMSO) were added and the magnitude of the force relaxation was recorded. The magnitude of relaxation was normalized to the magnitude of the pCa 6.7-induced force, which was assumed to be 100%.
The dependence of the force developed in response to different Ca2+ concentrations was determined by activating the strips by increasing the Ca2+ concentration from pCa > 8 to a higher Ca2+ concentration. The muscle strips were pre-incubated with the inhibitors (30 µM SL0101 or 30 µM quercitrin) or with corresponding concentrations of DMSO for 15 min in relaxing (G1) solution and then stimulated with Ca2+-activating solutions. The magnitude of the force response was measured in millinewtons.
3. Results and discussion
3.1. The binding of quercitrin to mRSK2NTKD
Using isothermal titration calorimetry (ITC), we determined that quercitrin binds to mRSK2NTKD with a K d of 5.8 µM (Fig. 2 ▶). This indicates a somewhat lower affinity compared with SL0101, for which a value of 2.9 µM was determined by us previously under comparable conditions (Utepbergenov et al., 2012 ▶). However, the low-micromolar K d shows that quercitrin readily forms a complex with mRSK2NTKD and strongly suggests that it will inhibit full-length wild-type RSK2.
Figure 2.

Thermodynamics of the binding of quercitrin (a) and SL0101 (b) to the N-terminal domain of mouse RSK2. The upper panels show raw titration data, while the lower panels show integrated titration curves and the final values of the determined thermodynamic parameters. The data for SL0101 are included for comparison and were originally determined and published in Utepbergenov et al. (2012 ▶).
3.2. Crystal structure of the quercitrin–mRSK2NTKD complex
The structure of the complex was solved by molecular replacement using the BALBES pipeline (Long et al., 2008 ▶), which utilized a version of the PDB that contained the AMPPNP–RSK2NTKD complex (Malakhova et al., 2009 ▶) but not the SL0101–RSK2NTKD complex (Utepbergenov et al., 2012 ▶), which was deposited after this work had been completed. Although the present structure is nearly isomorphous to that of mRSK2NTKD with bound SL0101, the independent structure solution and refinement avoided the introduction of any bias from the SL0101 complex. Data-collection and refinement statistics are listed in Table 1 ▶. In general terms, the overall structure is almost indistinguishable from the atomic models of the complexes with SL0101 and afzelin (Fig. 3 ▶). The quercitrin molecule is lodged within the ATP-binding site between the N- and C-terminal lobes of mRSK2NTKD in a manner similar to that observed for SL0101 (Utepbergenov et al., 2012 ▶) but different from that expected based on the available data on free flavonol binding.
Table 1. Data-collection and refinement statistics for the RSK2–quercitrin complex.
Data from one native crystal were used for structure determination. Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 98.08, b = 40.69, c = 83.26, β = 114.3 |
| Resolution range (Å) | 30.00–1.80 (1.84–1.80) |
| No. of unique reflections | 28004 (1389) |
| R merge † | 0.117 (0.303) |
| 〈I/σ(I)〉 | 15.2 (4.65) |
| Completeness (%) | 99.8 (100) |
| Multiplicity | 4.3 (3.8) |
| Refinement | |
| Resolution range (Å) | 25.30–1.80 (1.84–1.80) |
| No. of reflections | 27492 |
| R work/R free ‡ | 0.17/0.20 |
| No. of atoms | |
| Protein (non-H) | 2266 |
| Inhibitor (all atoms) | 52 |
| Water | 260 |
| B factors (Å2) | |
| Protein | 37.8 |
| Inhibitor | 29.9 |
| Water | 45.4 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.98 |
R
merge = 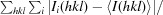
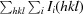 , where Ii(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of reflection hkl.
The crystallographic R factor R = 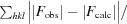
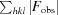 ; R
free =
, where all reflections belong to a test set of randomly selected data.
; R
free =
, where all reflections belong to a test set of randomly selected data.
Figure 3.
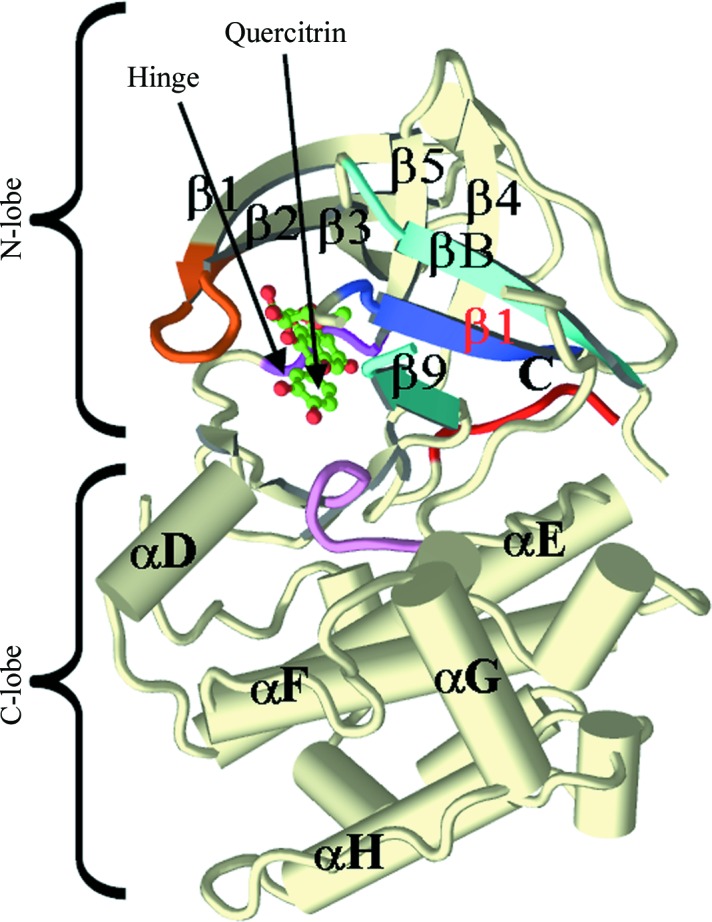
Overview of the crystal structure of the complex of quercitrin with the N-terminal kinase domain of mouse RSK2. All main secondary-structure elements, as well as the N- and C-lobes, are labeled. The Gly-rich loop is colored orange, the β-strands of the RSK-specific N-lobe sheet are shown in different shades of blue and the catalytic loop is colored purple. The hinge motif and the bound quercitrin are labeled.
3.2.1. General aspects of the binding of SL0101 and quercitrin to mRSK2NTKD
The adenine moiety of ATP is recognized by protein kinases through a conserved ‘three-point’ mechanism that in RSK2 involves a three-residue segment, Asp148-Phe149-Leu150, in the hinge region (Fig. 4 ▶ a). The six-membered ring of adenine is locked into position by the following hydrogen bonds: the 6-amino group donates a hydrogen bond to the main-chain carbonyl of the N-terminal hinge residue (Asp148 in RSK2), N1 accepts a hydrogen bond from the main-chain amide of the C-terminal residue (Leu150 in RSK2) and the heterocyclic carbon (C2) is involved in a C—H⋯O interaction with the carbonyl of the same amino acid. N3 of adenine is solvated, but does not interact directly with the protein.
Figure 4.
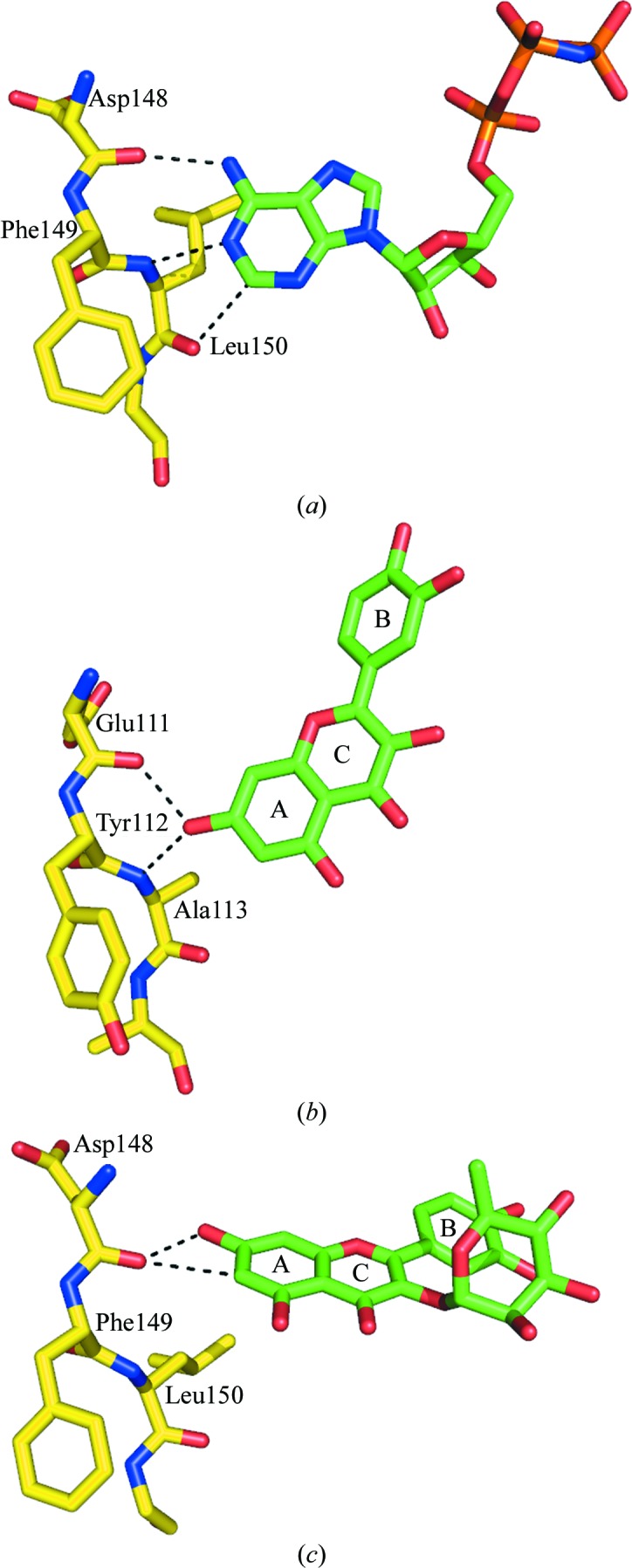
A comparison of the binding modes of adenine and flavonols to protein kinases. (a) The binding mode of the adenine moiety of AMPPNP to the hinge region of mRSK2NTKD (PDB entry 3g51; Malakhova et al., 2009 ▶). (b) The binding mode of quercetin to the hinge of protein kinase 17B (PDB entry 3lm5; Structural Genomics Consortium, unpublished work). (c) The binding mode of SL0101 to the hinge of mRSK2NTKD (PDB entry 3ubd; Utepbergenov et al., 2012 ▶). The binding mode of quercitrin reported in this study is identical to that of SL0101.
The binding mode of free flavonols, which typically mimic adenine, is exemplified by the structure of the Ser/Thr kinase 17B with bound quercetin (Fig. 4 ▶ b). The 7-hydroxyl positions itself to engage both the carbonyl of the N-terminal hinge residue and the amide of the C-terminal amino acid, both of which are engaged in the interaction with adenine. The C-terminal carbonyl of the hinge motif is in this case without a hydrogen-bonding partner. Nevertheless, the AC aromatic rings of the flavonol occupy almost exactly the equivalent space as the adenine of ATP.
In contrast to this stereochemistry, in the complexes of mRSK2NTKD with SL0101 and quercitrin the AC aromatic system of the inhibitor is rotated and shifted owing to the conformational change in the protein, so that the interaction of the 7-OH group with the N-terminal amide is disrupted (Fig. 4 ▶ c). Instead, the N-terminal carbonyl engages both the 7-OH and the aromatic C(6)—H group. Importantly, there are no other specific hydrogen bonds between the protein and the AC heterocycle. Thus, the AC system still makes contact with the hinge region of the kinase, but because the two lobes of the protein are twisted with respect to one another compared with the complexes with ATP or free flavonols (Utepbergenov et al., 2012 ▶), the orientation of the flavonol moiety in the cleft between the lobes is quite different from that of the canonical ATP binding.
As described for the SL0101 complex (Utepbergenov et al., 2012 ▶), the reorganization of the interface between the N- and C-lobes of mRSK2NTKD, which occurs as a consequence of the binding of the inhibitor, generates a hydrophobic pocket lined with 11 amino acids (i.e. Ile50, Ile52, Phe79, Leu102, Val131, Leu147, Leu150, Leu155, Leu200, Phe212 and Leu214), the surface of which is quite complementary to the surface of SL0101. The complex with quercitrin reveals an identical architecture for this pocket, with quercitrin bound in a very similar fashion, burying nearly 90% of its total solvent-accessible surface of ∼600 Å2 (Fig. 5 ▶). An interesting aspect of this binding mode of SL0101 and quercitrin to mRSK2NTKD is the conformation of the inhibitor bound to the protein. Owing to the presence of the carbohydrate at the 3-hydroxyl position, SL0101 and quercitrin have three degrees of conformational freedom, defined by the three dihedral angles ω, ϕ and ψ (see Fig. 1 ▶). The ω angle [C(3)—C(2)—C(1′)—C(2′)] defines the twist of the phenyl (B) ring with respect to the AC heterocyclic system, whereas the ϕ and ψ angles define the conformation around the glycosidic linkage. Although the structure of unbound SL0101 is not available, the crystal structure of free quercitrin is known (Jiang et al., 2009 ▶) and can be readily compared with that reported in our present study (Fig. 6 ▶ a). Clearly, the conformation of quercitrin bound to RSK2 (Fig. 6 ▶ b) is very different from that described for the unbound compound.
Figure 5.

The quercitrin molecule nested in the binding pocket between the N- and C-lobes of mRSK2NTKD. Quercitrin is shown using van der Waals spheres (oxygen in red and carbon in green); the protein is depicted as a surface representation. The three key residues making close contacts with quercitrin (and also depicted in Fig. 6 ▶) are shown as sticks and are colored and labeled.
Figure 6.

The structure of quercitrin unbound and bound to mRSK2NTKD. (a) Free quercitrin (CCDC code 698519), shown with H atoms not visualized and with the three dihedral conformational angles shown and labeled. (b) The conformation of quercitrin bound to mRSK2NTKD, shown along with an OMIT electron-density map contoured at 2σ; the values of the dihedral angles are shown. (c) A diagrammatic representation of the differences in the stereochemistry of bound quercitrin compared with bound SL0101.
Calculations of the free energies of unbound and bound quercitrin (after energy minimization of bond distances and angles involving H atoms only) indicate that free quercitrin has a lower energy by 2.490 kcal mol−1, a significant margin that is consistent with strain in the protein-bound state. The same is very likely to be true for SL0101. We believe that the bioactive conformation of quercitrin does not correspond to its global energy-minimum conformation in its free state. A systematic search of the conformations of quercitrin generated, after energy minimization, 14 unique low-energy conformations (Supplementary Figs. S1 and S2 and Table S11) from the 363 initial conformers. The lowest energy conformation of these (CNF13) is the global vacuum minimum. Not only does the bound conformation optimize to a local minimum (CNF2, with a relative energy of 2.72 kcal mol−1), which suggests that the ligand is highly flexible, but its strain energy within the site is a further 8.192 kcal mol−1 higher than the local minimum.
In both structures the phenyl (B) ring is twisted out of the plane of the AC system, albeit in opposite directions. It has been reported that in flavonoids which lack the 3-hydroxyl the three rings are nearly coplanar and the ω angle is close to 0° (Yang et al., 2008 ▶; Sharma et al., 2007 ▶). The presence of the 3-hydroxyl slightly increases the deviation from planarity, as shown in the two independent crystal structures of quercetin (Rossi et al., 1986 ▶; Jin et al., 1990 ▶). Moreover, the magnitude of the rotation of the B ring increases in proportion to the size of the group attached to C(3), so in the structure of free quercitrin the ω angle is −29.3° (Jiang et al., 2009 ▶). In bound quercitrin the ω angle is 15.4°, so that the B ring is twisted in the opposite direction from that observed in free quercitrin owing to the different disposition of the α-l-rhamnose moiety defined by the two dihedral angles of the glycosidic linkage, ϕ and ψ [C(1′′)—O(7)—C(3)—C(2) and C(3)—O(7)—C(1′′)—C(2′′), respectively]. In free quercitrin the ϕ and ψ angles are −70.9° and 168.0°, respectively, resulting in an ‘open’ clover-like structure in which l-rhamnose, the B ring and the AC system make no close van der Waals contacts. In contrast, the bound quercitrin molecule has a conformation in which the carbohydrate packs snugly against the phenyl (B) ring, with the ϕ and ψ angles assuming values of 130.3° and −177.4°, respectively. The largest conformational change in quercitirin resulting from its binding to the protein is a major swing of the carbohydrate moiety around the ϕ angle by ∼160°, placing it in van der Waals proximity of the B ring and causing it to rotate counterclockwise with respect to the AC plane.
The bound SL0101 shows the same features, with some notable differences in the dihedral angles. The ω angle in SL0101 is 29.0°, a significantly larger twist than in quercitrin. The ϕ and ψ angles of the glycosidic linkage are 120.1° and −172.5°, respectively, corresponding to rotations of −10.2° and 4.9° compared with quercitrin. We will now discuss the significance of these differences.
3.2.2. Differences between the binding modes of SL0101 and quercitrin
The most conspicuous difference, which is clearly visible in the electron density, between the two inhibitors bound to RSK2NTKD is the expected presence of an additional hydroxyl in the C(3′) position of the B ring in quercitrin (Fig. 6 ▶ b). Although in principle the phenyl ring can flip 180°, leading to two alternate positions for the hydroxyl, only one conformation is observed in the crystal structure with the hydroxyl positioned proximally to the l-rhamnose moiety. The alternative conformation (as observed, for example, in free quercetin) is not sterically possible, as it would bring the additional hydroxyl into a steric clash with the Cγ2 methyl of Thr210. The additional C(3′) hydroxyl of quercitrin comes into close contact with the side chain of Leu155. This short distance rationalizes the observed difference in the ω angle, which decreases to 15.4° from 29.0° in bound SL0101 as the B ring is forced to rotate away from Leu155 (Fig. 6 ▶ c). This rotation in turn exerts direct pressure on the l-rhamnose. As the B ring of quercitrin is forced to rotate by ∼15° owing to a steric clash caused by the C(3′) hydroxyl, it pushes on the l-rhamnose moiety, causing it to swing away, resulting in an ∼−10° rotation around the ϕ angle and an additional ∼5° rotation around the ψ angle. This rearrangement brings l-rhamnose into close contact with Leu102. Thus, the additional C(3′) hydroxyl on the B ring in quercitrin introduces a steric clash with Leu155, which is consistent with the lower affinity of quercitrin for RSK2NTKD compared with SL0101. The close contacts cannot be easily dissipated because SL0101 and quercitrin are already bound in a strained conformation packing the l-rhamnose against the B ring.
3.3. Quercitrin as an alternative to SL0101 in a biological assay
We recently showed that SL0101 inhibits the RSK kinase in smooth muscle and consequently down-regulates the contractile force (Artamonov et al., submitted). We therefore wondered whether quercitrin can be used in this biological assay with the same effect as SL0101.
Briefly, smooth-muscle (SM) cells contract in response to Ca2+ influx through membrane channels, as well as Ca2+ release from the sarcoplasmic reticulum (SR). Contraction is induced when Ca2+ binds to the regulatory protein calmodulin, which in its metal-bound form interacts with and activates the myosin light-chain kinase (MLCK), which in turn phosphorylates the 20 kDa regulatory myosin light chain (RLC20) on Ser19 with concomitant activation of the ATPase activity of myosin (Somlyo & Somlyo, 1994 ▶). Conversely, dephosphorylation of RLC20 by the RLC20 phosphatase (MLCP) inhibits contraction and induces relaxation (Hartshorne et al., 2004 ▶; Ito et al., 2004 ▶). This basic cycle is modulated in a complex and often tissue-specific way by phenomena which are both Ca2+-dependent and Ca2+-independent, with the latter being defined as ‘Ca2+ sensitization’ (Kitazawa et al., 1989 ▶). This agonist-activated pathway is mediated by select G-protein-coupled receptors (GPCRs) which activate several guanine nucleotide-exchange factors for RhoA, which in turn binds to and activates the ROCK kinase with the ultimate effect of phosphorylation and inhibition of MLCP (Wirth et al., 2008 ▶; Momotani & Somlyo, 2012 ▶).
Of particular interest in the field is the potential role in SM contractility of additional protein kinases which may directly or indirectly act on MLCK, MLCP or both. Several protein kinases have been implicated, including ZIPK (Moffat et al., 2011 ▶), integrin-linked kinase (ILK; Wilson et al., 2005 ▶) and PAK kinase (Wirth et al., 2003 ▶). Recently, we showed that p90 ribosomal S6 kinase (most likely the RSK2 isoform) is physiologically active in SM and mediates in part both the Ca2+-dependent and the independent contractile response through phosphorylation of RLC20 and phosphorylation of the MYPT1 subunit of MLCP, leading to inhibition of phosphatase activity and increased contractile force (Artamonov et al., submitted). Therefore, we decided to use these contractility assays to test whether quercitrin can replace SL0101, despite its lower apparent binding constant.
In the first experiment, we determined that in the contractility assays, which were carried out with rabbit pulmonary artery permeabilized with S. aureus α-toxin, the use of 30 µM quercitrin increased the Ca2+ concentration threshold for the onset of contraction and significantly reduced the maximal Ca2+-induced contractile force (Fig. 7 ▶). These effects mimicked those generated by SL0101, but the magnitude of the response was greater. We then carried out an experiment to determine the IC50 for quercitrin. We found that quercitrin had an IC50 value of 17.5 µM (Fig. 8 ▶). Determination of the precise IC50 value for SL0101 under the same experimental conditions proved difficult owing to limited solubility of the compound compared with quercitrin. Under the conditions of the ex vivo contractility assay, in which the ATP concentration was 4.5 mM, the IC50 values are expected to be higher than under standard conditions (e.g. 10 µM ATP, as typically used in RSK assays).
Figure 7.

The RSK2 inhibitors quercitrin and SL0101 decrease the maximal force at each intracellular Ca2+ concentration (pCa). The significance of the difference between curves was determined using two-way analysis of variance. Corresponding column factor P values are shown on each graph; n = 4–9.
Figure 8.

A dose-response curve for quercitrin in α-toxin-permeabilized rabbit pulmonary artery smooth muscle. The significance of the difference between curves was determined using two-way analysis of variance. P < 0.0001 versus diluent control; n = 3–4.
4. Conclusions
As a selective RSK inhibitor, SL0101 has proven to be a very useful tool in cell biology. Among other uses, it has been used to demonstrate that RSK2 kinase controls cell survival through its ability to regulate the formation of RNA granules during stress (Eisinger-Mathason et al., 2008 ▶). It has also been used to show that RSK regulates cell fate in the human breast ductal network (Pasic et al., 2011 ▶) and that it phosphorylates Y-box protein-1 on Ser102 in basal-like breast-cancer cells (Stratford et al., 2008 ▶). When used in combination with a PKCζ pseudosubstrate, SL0101 completely abrogated ANG-II-induced RSK2-mediated cell proliferation (Godeny & Sayeski, 2006 ▶). The compound inhibited glucose uptake in 3T3-L1 adipocytes, which occurs, at least in part, through RSK-dependent phosphorylation of the Na+/H+-exchanger NHE1 (Chen & Mackintosh, 2009 ▶).
Despite its obvious utility, SL0101 is an esoteric compound that has so far only been identified in nature in a single Amazonian plant species, making the commercially available synthetic reagent expensive ($250 per milligram). On the other hand, its close relative quercitrin is abundant and inexpensive (∼$2 per milligram). It is found in, among others, Tartary buckwheat (Fagopyrum tataricum), Persian berries and oak trees such as the North American white oak and black oak and the European red oak. The compound has an interesting history. Quercetin is the active ingredient of quercitrin and was discovered in 1771 by an American physician, Dr Edward Bancroft (1744–1821). It was used as a popular dyeing substance and was originally obtained from the Eastern black oak (the other ingredient is quercitannic acid). Today, pure quercitrin is readily available from many manufacturers. Interestingly, there have been a number of studies suggesting anti-inflammatory and antimicrobial effects of quercitrin and even cytoprotective effects against ultraviolet B radiation (Yang et al., 2012 ▶). However, quercitrin ingested with a normal diet is unlikely to have any physiological effects, as it is not absorbed in the gut owing to the presence of l-rhamnose, which cannot be hydrolyzed by any of the human glycosidases. A fraction of the glycoside is probably hydrolyzed by the gut microbiota, allowing the aglycone quercetin to be absorbed into the bloodstream.
In this study, we show that quercitrin binds to the N-terminal domain of RSK2 with slightly lower affinity than SL0101, probably owing to steric effects caused by the introduction of an additional hydroxyl on the B ring. Like SL0101, quercitrin is bound in a strained conformation. We recently suggested that SL0101 binds to RSK through an induced-fit mechanism, so that the encounter complex contains a canonical tertiary structure of the kinase (Utepbergenov et al., 2012 ▶). It is also quite probable that the encounter complex contains a low-energy conformer of SL0101 (or quercitrin) and that the conformer observed in the crystal structure is the result of simultaneous stereochemical rearrangement in the protein and the inhibitor by an as yet uncharacterized mechanism. The atomic model of the complex of quercitrin with RSK2NTKD shows that in its conformational relaxed state quercitrin cannot enter the binding site in the enzyme owing to a significant steric clash.
We also show evidence that quercitrin can be used in cell biology as an inexpensive SL0101 surrogate. In our smooth-muscle contractility assays, quercitrin successfully reproduced the effects of SL0101 and was even more effective owing to its higher solubility. While further studies are required to establish the selectivity of quercitrin, the crystal structure suggests that the compound will show a selectivity similar to that reported for SL0101 and that it can be used as its surrogate.
Supplementary Material
PDB reference: N-terminal kinase domain of murine RSK2, complex with quercitrin, 4gue
Supplementary material file. DOI: 10.1107/S0907444912045520/en5521sup1.pdf
Acknowledgments
This research was supported by the University of Virginia Cancer Center through the NCI Cancer Center Support Grant P30 CA44579 and in part by the National Institutes of Health (R01 GM086457 to ZSD and AVS). The use of the Advanced Photon Source (APS) was supported by the US Department of Energy, Office of Science and Office of Basic Energy Sciences under contract No. W-31-109-Eng-38. Supporting institutions of SER-CAT may be found at http://www.ser-cat.org/members.html.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: EN5521).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Anjum, R. & Blenis, J. (2008). Nature Rev. Mol. Cell Biol. 9, 747–758. [DOI] [PubMed]
- Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H., Klevernic, I., Arthur, J. S., Alessi, D. R. & Cohen, P. (2007). Biochem. J. 408, 297–315. [DOI] [PMC free article] [PubMed]
- Boly, R., Gras, T., Lamkami, T., Guissou, P., Serteyn, D., Kiss, R. & Dubois, J. (2011). Int. J. Oncol. 38, 833–842. [DOI] [PubMed]
- Calderón-Montaño, J. M., Burgos-Morón, E., Pérez-Guerrero, C. & López-Lázaro, M. (2011). Mini Rev. Med. Chem. 11, 298–344. [DOI] [PubMed]
- Chen, S. & Mackintosh, C. (2009). Cell. Signal. 21, 1984–1993. [DOI] [PubMed]
- Cho, Y.-Y., Yao, K., Pugliese, A., Malakhova, M. L., Bode, A. M. & Dong, Z. (2009). Cancer Res. 69, 4398–4406. [DOI] [PMC free article] [PubMed]
- Clark, D. E., Errington, T. M., Smith, J. A., Frierson, H. F., Weber, M. J. & Lannigan, D. A. (2005). Cancer Res. 65, 3108–3116. [DOI] [PubMed]
- Cohen, S. X., Ben Jelloul, M., Long, F., Vagin, A., Knipscheer, P., Lebbink, J., Sixma, T. K., Lamzin, V. S., Murshudov, G. N. & Perrakis, A. (2008). Acta Cryst. D64, 49–60. [DOI] [PMC free article] [PubMed]
- Cuadrado, A. & Nebreda, A. R. (2007). Cancer Cell, 12, 187–189. [DOI] [PubMed]
- Eisinger-Mathason, T. S., Andrade, J., Groehler, A. L., Clark, D. E., Muratore-Schroeder, T. L., Pasic, L., Smith, J. A., Shabanowitz, J., Hunt, D. F., Macara, I. G. & Lannigan, D. A. (2008). Mol. Cell, 31, 722–736. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Godeny, M. D. & Sayeski, P. P. (2006). Am. J. Physiol. Cell Physiol. 291, C1308–C1317. [DOI] [PubMed]
- Goettert, M., Schattel, V., Koch, P., Merfort, I. & Laufer, S. (2010). Chembiochem, 11, 2579–2588. [DOI] [PubMed]
- Hartshorne, D. J., Ito, M. & Erdödi, F. (2004). J. Biol. Chem. 279, 37211–37214. [DOI] [PubMed]
- Holder, S., Zemskova, M., Zhang, C., Tabrizizad, M., Bremer, R., Neidigh, J. W. & Lilly, M. B. (2007). Mol. Cancer Ther. 6, 163–172. [DOI] [PubMed]
- Horiuti, K. (1988). J. Physiol. 398, 131–148. [DOI] [PMC free article] [PubMed]
- Hou, D.-X. & Kumamoto, T. (2010). Antioxid. Redox Signal. 13, 691–719. [DOI] [PubMed]
- Ito, M., Nakano, T., Erdodi, F. & Hartshorne, D. J. (2004). Mol. Cell. Biochem. 259, 197–209. [DOI] [PubMed]
- Jiang, R.-W., Wang, Y., Gao, H., Zhang, D.-M. & Ye, W.-C. (2009). J. Mol. Struct. 920, 383–386.
- Jin, G.-Z., Yamagata, Y. & Tomita, K. (1990). Acta Cryst. C46, 310–313.
- Jones, S. W., Erikson, E., Blenis, J., Maller, J. L. & Erikson, R. L. (1988). Proc. Natl Acad. Sci. USA, 85, 3377–3381. [DOI] [PMC free article] [PubMed]
- Kang, S., Dong, S., Gu, T.-L., Guo, A., Cohen, M. S., Lonial, S., Khoury, H. J., Fabbro, D., Gilliland, D. G., Bergsagel, P. L., Taunton, J., Polakiewicz, R. D. & Chen, J. (2007). Cancer Cell, 12, 201–214. [DOI] [PMC free article] [PubMed]
- Kang, S. et al. (2009). Mol. Cell. Biol. 29, 2105–2117. [DOI] [PMC free article] [PubMed]
- Kang, S. et al. (2010). J. Clin. Invest. 120, 1165–1177. [DOI] [PMC free article] [PubMed]
- Kim, W., Yang, H. J., Youn, H., Yun, Y. J., Seong, K. M. & Youn, B. (2010). J. Radiat. Res. 51, 285–296. [DOI] [PubMed]
- Kitazawa, T., Kobayashi, S., Horiuti, K., Somlyo, A. V. & Somlyo, A. P. (1989). J. Biol. Chem. 264, 5339–5342. [PubMed]
- Lee, K. M., Lee, D. E., Seo, S. K., Hwang, M. K., Heo, Y.-S., Lee, K. W. & Lee, H. J. (2010). Carcinogenesis, 31, 1338–1343. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Lu, W., Liu, X., Cao, X., Xue, M., Liu, K., Zhao, Z., Shen, X., Jiang, H., Xu, Y., Huang, J. & Li, H. (2011). J. Med. Chem. 54, 3564–3574. [DOI] [PubMed]
- Maekawa, N., Abe, J., Shishido, T., Itoh, S., Ding, B., Sharma, V. K., Sheu, S.-S., Blaxall, B. C. & Berk, B. C. (2006). Circulation, 113, 2516–2523. [DOI] [PubMed]
- Malakhova, M., Kurinov, I., Liu, K. D., Zheng, D., D’Angelo, I., Shim, J.-H., Steinman, V., Bode, A. M. & Dong, Z. G. (2009). PLoS One, 4, e8044. [DOI] [PMC free article] [PubMed]
- Maloney, D. J. & Hecht, S. M. (2005). Org. Lett. 7, 1097–1099. [DOI] [PubMed]
- Maron, D. J. (2004). Curr. Atheroscler. Rep. 6, 73–78. [DOI] [PubMed]
- Mirmohammadsadegh, A., Mota, R., Gustrau, A., Hassan, M., Nambiar, S., Marini, A., Bojar, H., Tannapfel, A. & Hengge, U. R. (2007). J. Invest. Dermatol. 127, 2207–2215. [DOI] [PubMed]
- Moffat, L. D., Brown, S. B., Grassie, M. E., Ulke-Lemée, A., Williamson, L. M., Walsh, M. P. & MacDonald, J. A. (2011). J. Biol. Chem. 286, 36978–36991. [DOI] [PMC free article] [PubMed]
- Momotani, K. & Somlyo, A. V. (2012). Trends Cardiovasc. Med. 22, 122–127 [DOI] [PMC free article] [PubMed]
- Neuhouser, M. L. (2004). Nutr. Cancer, 50, 1–7. [DOI] [PubMed]
- Nguyen, T. L. (2008). Anticancer Agents Med. Chem. 8, 710–716. [DOI] [PubMed]
- Nguyen, T. L., Gussio, R., Smith, J. A., Lannigan, D. A., Hecht, S. M., Scudiero, D. A., Shoemaker, R. H. & Zaharevitz, D. W. (2006). Bioorg. Med. Chem. 14, 6097–6105. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pasic, L., Eisinger-Mathason, T. S., Velayudhan, B. T., Moskaluk, C. A., Brenin, D. R., Macara, I. G. & Lannigan, D. A. (2011). Genes Dev. 25, 1641–1653. [DOI] [PMC free article] [PubMed]
- Pearce, L. R., Komander, D. & Alessi, D. R. (2010). Nature Rev. Mol. Cell Biol. 11, 9–22. [DOI] [PubMed]
- Rogers, J. C. & Williams, D. L. (1989). Biochem. Biophys. Res. Commun. 164, 419–425. [DOI] [PubMed]
- Romeo, Y. & Roux, P. P. (2011). Expert Opin. Ther. Targets, 15, 5–9. [DOI] [PubMed]
- Rossi, M., Rickles, L. F. & Halpin, W. A. (1986). Bioorg. Chem. 14, 55–69.
- Sharma, D., Gupta, V. K., Brahmachari, G., Mondal, S. & Gangopadhyay, A. (2007). Bull. Mater. Sci. 30, 469–475.
- Sheffield, P., Garrard, S. & Derewenda, Z. (1999). Protein Expr. Purif. 15, 34–39. [DOI] [PubMed]
- Sicheri, F., Moarefi, I. & Kuriyan, J. (1997). Nature (London), 385, 602–609. [DOI] [PubMed]
- Smith, J. A., Poteet-Smith, C. E., Xu, Y., Errington, T. M., Hecht, S. M. & Lannigan, D. A. (2005). Cancer Res. 65, 1027–1034. [PubMed]
- Somlyo, A. P. & Somlyo, A. V. (1994). Nature (London), 372, 231–236. [DOI] [PubMed]
- Stratford, A. L. & Dunn, S. E. (2011). Expert Opin. Ther. Targets, 15, 1–4. [DOI] [PubMed]
- Stratford, A. L., Fry, C. J., Desilets, C., Davies, A. H., Cho, Y. -Y., Li, Y., Dong, Z., Berquin, I. M., Roux, P. P. & Dunn, S. E. (2008). Breast Cancer Res. 10, R99. [DOI] [PMC free article] [PubMed]
- Suh, Y., Afaq, F., Khan, N., Johnson, J. J., Khusro, F. H. & Mukhtar, H. (2010). Carcinogenesis, 31, 1424–1433. [DOI] [PMC free article] [PubMed]
- Utepbergenov, D., Derewenda, U., Olekhnovich, N., Szukalska, G., Banerjee, B., Hilinski, M. K., Lannigan, D. A., Stukenberg, P. T. & Derewenda, Z. S. (2012). Biochemistry, 51, 6499–6510. [DOI] [PMC free article] [PubMed]
- Walker, E. H., Pacold, M. E., Perisic, O., Stephens, L., Hawkins, P. T., Wymann, M. P. & Williams, R. L. (2000). Mol. Cell, 6, 909–919. [DOI] [PubMed]
- Williams, R. J., Spencer, J. P. & Rice-Evans, C. (2004). Free Radic. Biol. Med. 36, 838–849. [DOI] [PubMed]
- Wilson, D. P., Sutherland, C., Borman, M. A., Deng, J. T., MacDonald, J. A. & Walsh, M. P. (2005). Biochem. J. 392, 641–648. [DOI] [PMC free article] [PubMed]
- Wirth, A., Benyó, Z., Lukasova, M., Leutgeb, B., Wettschureck, N., Gorbey, S., Orsy, P., Horváth, B., Maser-Gluth, C., Greiner, E., Lemmer, B., Schütz, G., Gutkind, J. S. & Offermanns, S. (2008). Nature Med. 14, 64–68. [DOI] [PubMed]
- Wirth, A., Schroeter, M., Kock-Hauser, C., Manser, E., Chalovich, J. M., De Lanerolle, P. & Pfitzer, G. (2003). J. Physiol. 549, 489–500. [DOI] [PMC free article] [PubMed]
- Yang, H.-M., Ham, Y.-M., Yoon, W.-J., Roh, S. W., Jeon, Y.-J., Oda, T., Kang, S.-M., Kang, M.-C., Kim, E.-A., Kim, D. & Kim, K.-N. (2012). J. Photochem. Photobiol. B, 114, 126–131. [DOI] [PubMed]
- Yang, H.-B., Wang, Y.-C., Zhang, Z.-T. & Chang, Y. (2008). Turk. J. Chem. 32, 87–95.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: N-terminal kinase domain of murine RSK2, complex with quercitrin, 4gue
Supplementary material file. DOI: 10.1107/S0907444912045520/en5521sup1.pdf