

Abstract
Prolonged morphine treatment increases pain sensitivity in many patients. Enhanced spinal Substance P release is one of the adaptive changes associated with sustained opioid exposure. In addition to pain transmitting second order neurons, spinal microglia and astrocytes also express functionally active Tachykinin NK1 (Substance P) receptors. In the present work we investigated the role of glial Tachykinin NK1 receptors in morphine withdrawal-mediated spinal microglia and astrocyte activation. Our data indicate that intrathecal co-administration (6 days, twice daily) of a selective Tachykinin NK1 receptor antagonist (N-acetyl-l-tryptophan 3,5-bis(trifluoromethyl)benzylester (L-732,138; 20 μg/injection) attenuates spinal microglia and astrocyte marker and pro-inflammatory mediator immunoreactivity as well as hyperalgesia in morphine-withdrawn rats. Furthermore, covalent linkage of the opioid agonist with a Tachykinin NK1 antagonist pharmacophor yielded a bivalent compound that did not augment spinal microglia or astrocyte marker or pro-inflammatory mediator immunoreactivity and did not cause paradoxical pain sensitization upon drug withdrawal. Thus, bivalent opioid/Tachykinin NK1 receptor antagonists may provide a novel paradigm for long-term pain management.
Keywords: opioid-induced hyperalgesia, spinal glia, Tachykinin NK1 receptor, Tachykinin NK1 receptor antagonist
1. Introduction
Morphine and other opiate agonists provide efficient relief from severe acute pain. The use of opiates for the management of chronic pain is more problematic, since long-term opiate analgesic administration leads to antinociceptive tolerance (Freye et al., 2003) and paradoxically, increases the sensitivity of experimental animals (Ossipov et al., 2003) as well as human patients (Simonnet and Rivat, 2003) to mildly painful (opioid-induced hyperalgesia and innocuous (allodynia) stimuli. It was suggested earlier that such paradoxical pain sensitization contributes to apparent antinociceptive tolerance (Mao et al., 1994).
In the past decade it became increasingly clear that resident non-neuronal cells - glia - within the central nervous system (CNS) have a crucial role in the regulation of pain sensitivity (Raghavendra et al., 2003, Watkins et al., 2005). Accordingly, inhibition of glial activation markedly attenuates neural injury and inflammation-mediated pain sensitization (Raghavendra et al., 2003). Thus, regulation of glial activation provides an exciting novel target for analgesic drug development (Watkins et al., 2005). Interestingly, similar to inflammation and neuropathies, sustained opioid analgesic treatment was also found to activate CNS glia (Watkins et al., 2005, Watkins et al., 2007)) and glial inhibitors were able to attenuate sustained morphine-mediated paradoxical pain sensitization (Mika et al., 2007). Thus, it was suggested that spinal glial activation might play a role in sustained opioid-mediated pain facilitation (Watkins et al., 2005, Watkins et al., 2007, Mika et al., 2007).
Enhanced spinal excitatory pain neurotransmitter (such as Calcitonin Gene-related Peptide (CGRP) and Substance P) synthesis and/or release is one of the major adaptive changes associated with sustained opioid exposure (King et al., 2005). Substance P release from the peptidergic unmyelinated sensory neurons (C-fibres) is thought to activate Tachykinin NK1 receptors on second order neurons leading to acute pain signal transmission (Allen et al., 1997). Interestingly, in addition to pain transmitting second order neurons, spinal microglia and astrocytes also express functionally active Tachykinin NK1 receptors (Marriott, 2004; Pocock and Kettenmann, 2007). Activation of Tachykinin NK1 receptors in cultured microglia was shown to lead to p38 MAP kinase activation (Fiebich et al., 2000, Svensson et al., 2005) and augmented pro-inflammatory modulator synthesis (Fiebich et al., 2000; Marriott, 2004) indicating that glial Tachykinin NK1 receptors regulate neuroimmune responses. In the present work we investigated the hypothesis that attenuation of spinal microglial and/or astrocyte Tachykinin NK1 receptor activation will attenuate sustained opioid agonist-mediated glial activation and thus will prevent the development of opioid-induced hyperalgesia. Our data indicate that intrathecal (i.th.) Tachykinin NK1 receptor antagonist co-administration attenuates sustained intraperitoneal (i.p.) morphine-mediated upregulation of spinal microglia and astrocyte marker immunoreactivity and prevents morphine withdrawal-mediated thermal hyperalgesia and tactile allodynia in rats. Covalent linkage of the opioid agonist pharmacophor with a Tachykinin NK1 antagonist yielded a bivalent compound that did not augment spinal microglia or astrocyte marker immunoreactivity and did not cause paradoxical pain sensitization upon drug withdrawal.
2. Materials and methods
2.1. Animals
Adult (200–225 g) male Sprague-Dawley rats (Harlan Sprague-Dawley, Indianapolis, IN) were kept in a climate-controlled room on a 12 h light/dark cycle with food and water available ad libitum. Handling, care, maintenance and testing of the animals were performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines for the handling and use of laboratory animals. The experimental protocol was approved by the Animal Care and Use Committee of the University of Arizona.
2.2. Intrathecal catheter implantation
All animals underwent intrathecal catheterization, followed by a three-day recovery period. For catheter implantation, the animals were anesthetized by ketamine-xylazine (100 mg/kg, i.p) and were implanted with intrathecal (i.th.) catheters (8 cm polyethylene-10 tubing) so that the catheter terminated in the lumbar region of the spinal cord (Yaksh and Rudy, 1976). The animals were housed individually and allowed to recover for 7 days before any further experimental manipulations. Animals with signs of motor weakness or paralysis or with greater than 10% loss in body weight were excluded from further experimentation. Prior to drug treatment, the free flow of fluids through the catheters was verified by injecting saline solution. All the animals were subjected to baseline nociceptive tests prior to any drug intervention.
2.3. Drug administration
Long-term morphine administration was accomplished by intraperitoneal (i.p.) injection of morphine sulfate (5mg/kg/injection, NIDA, Bethesda, Maryland) to rats twice daily, for 6 days (saline-morphine group, n=6 animals). Separate groups of animals received intrathecal (i.th.) injections of the selective Tachykinin NK1 receptor antagonist, [N-acetyl-l-tryptophan 3,5-bis(trifluoromethyl)benzylester (L-732,138; 20 μg/5 μl/injection; Tocris, Ellisville, Missouri), twice daily for 6 days, together with either morphine (morphine− L-732,138 group, n=6) or saline (saline- L-732,138 group, n=6 animals). The dose of L-732,138 was selected based on dose-response relationship studies (unpublished observations); L-732138 was dissolved in a solution containing 10% DMSO and 90% 0.9 % saline (vehicle). Alternatively, a group of animals received twice daily intrathecal injections (20 μg/5 μl/injection) of a novel bivalent compound TY027 (H-Tyr-D-Ala-Gly-Phe-Met- Pro-Leu-Trp-NH-3, 5 Bn(CF3)2) provided by Dr. Victor J Hruby at the University of Arizona and synthesized as described by Yamamoto et al. (2007, 2008) along with saline (i.p) injections (TY027-saline group). In TY027, an opioid peptide pharmacophor (Tyr-DAla-Gly-Phe-Met) has been covalently coupled to a Tachykinin NK1 receptor antagonist pharmacophor (Leu-Trp-NHBn(CF3)2). TY027 was also dissolved in a solution containing 10% DMSO and 90% 0.9 % saline. The dose of TY027 was selected based on the studies of Largent-Milnes (2010a). Control animals received either the same volume of i.th. saline (saline-saline group) and/or vehicle (saline-vehicle group) injections. Animals in the morphine-saline and morphine-vehicle group received i.th. saline or vehicle injections, respectively. The animals receiving saline-vehicle injections exhibited pain thresholds similar to that of saline-saline control group. The animals receiving the morphine-vehicle injections exhibited pain thresholds similar to that of morphine-saline group. All the compounds were administered twice daily for 6 days. Behavioral (thermal hyperalgesia and tactile allodynia) assays were performed prior to any drug administration (basal values), and after 96 h withdrawal to allow for the complete drug clearance after the last drug injection.
2.4. Measurement of thermal hyperalgesia
The method of Hargreaves et al. (1988) was used to assess the sensitivity of rats to a mildly noxious thermal stimulus, as previously described (Gardell et al., 2002). Briefly, the animals (6 individual animals in each treatment group) were placed in a glass floor container and a radiant heat source was focused onto the plantar surface of their hind-paw. Paw withdrawal latencies in seconds were measured using a motion detector, before (baseline) drug administration and 96 h after the last drug injection. A maximal cutoff of 33 s was used to prevent tissue damage.
2.5. Measurement of mechanical allodynia
Paw withdrawal threshold in response to mild, normally innocuous tactile stimuli were determined by probing with von Frey filaments, as previously described (Gardell et al., 2002). The animals (6 individual animals in each treatment group) were placed in a plexi-glass container equipped with a wire mesh floor. The filaments (0.4 –15.1 gm)were applied perpendicularly to the plantar surface of the right hind paw of the rats. Paw withdrawal thresholds were measured by sequentially increasing and then decreasing the stimulus strength (“up-and-down” method). Paw withdrawal thresholds were calculated in grams using the Dixon non-parametric test and expressed as the mean paw withdrawal threshold ± S.E.M.
2.6. Immunohistochemistry
Immunohistochemistry was performed using a protocol modified from Gardell et al. (2002). Briefly, after behavioural testing, the rats that responded positively to the hyperalgesic test at 96 h withdrawal time point were selected from each group and were anesthetized with intraperitoneal ketamine-xylazine (100mg/kg) injections and transcardially perfused with 0.1 M PBS (pH 7.4) until the exudate ran clear and then with 10% formalin for 15 min. Lumbar (L) spinal cords were harvested. The tissues were fixed in 10% formalin solution overnight and cryoprotected with 20% sucrose in 0.1 M PBS. The fixed tissues were saturated with 30% sucrose in 0.1 M PBS solution and embedded in Tissue-Tek Optimal Cutting Temperature Compound (Sakura, Torrance, CA) and sliced in a cryostat at −20°C. Serial spinal cord sections (20μm each) were placed onto slides so that each slide contained an ordered series of sections from a lumbar spinal cord. The mounted lumbar spinal cord sections were extensively rinsed and blocked in 0.1 M PBS containing 10% goat serum, for 2 h, at room temperature. The sections were incubated with a mouse monoclonal anti-Glial Fibrillary Acidic Protein (GFAP) antiserum (1:2,000; Chemicon International, Temecula, CA) to label activated astrocytes or a mouse monoclonal anti-complement type 3 receptor (CR3/CD11b) antibody (OX-42) (1:2,000; Chemicon International, Temecula, CA) in 0.1 M Phosphate-buffered Saline (PBS) containing 2% goat serum and 0.3% Triton X-100 overnight at 4°C. The slides were subsequently washed and incubated (2 h) with an Alexa Fluor 594-conjugated goat anti-mouse secondary antibody (1:1000; Molecular Probes, Eugene, OR). After immunoreactions, the sections were rinsed and mounted in the Vectashield (Vector Laboratories) mounting medium. Fluorescence images were digitally captured using a Nikon (Tokyo, Japan) E800 fluorescence microscope, equipped with the appropriate standard filters. Images were acquired and analyzed using a Hamamatsu C5810 color CCD camera and its proprietary Image Processor Software (Hamamatsu, Bridgewater, NJ). The acquired images were processed using Adobe PhotoShop (Adobe Systems, San Jose, CA). For quantification of GFAP and OX-42 immunofluorescence, an area within the dorsal horn of the spinal cord of each treatment group was outlined and immunofluorescence intensities above background were quantified using NIH Image J software. The total Intensity of immunofluorescence above a predefined threshold for 4 pre-determined sections within the dorsal horn of each individual rat was averaged to provide a mean Intensity value for each rat. A total of 3 independent animals were used per treatment group for the quantification analysis.
2.7. Rat TNFα (Tumor Necrosis Factor α) ELISA assay
After drug treatment and behavioral tests, the animals were sacrificed, their spinal columns were hydraulically extruded with ice-cold saline and the lumbar cord was dissected on ice as described in Gardell et al. (2002). The tissue samples were immediately frozen in liquid nitrogen and stored at. On the day of the experiment, frozen (−80°C) lumbar spinal cord tissues were thawed and homogenized as previously described (Gardell et al., 2002). After centrifugation (10,000 × g, 30 min, 4°C) a TNFα ELISA assay (Thermo Scientific, rat TNFα ELISA kit; Cat#: ER3TNFA) was performed according to the manufacturer’s instructions. Briefly, the samples were diluted 1:1 in the standard diluent, loaded onto a 96 well plate (50 μg protein/well). After 1h incubation at room temperature, the samples were washed three times with a washing buffer and incubated with a biotinylated anti-rat TNFα antibody (1h, RT), followed by incubation with streptavidin-HRP (Horse Radish Peroxidfase) and a colorigenic substrate (TMB). Absorbance values were measured at 450 and 550 nm. Standard curves were constructed using synthetic TNFα solutions. Data were calculated using the standard curve and analyzed using a GrapPad Prism Software.
2.8. Data analysis
Thermal and tactile hypersensitivity data were analyzed by one-way ANOVA (post hoc: Newman-Keuls) using the GraphPad Prism 4.0 software. The mean intensity values of the pre determined immunofluorescent sections, as measured using NIH ImageJ software, were normalized and plotted using the GraphPad Prism 4.0 software. One sample t-tests were used to compare normalized mean intensity values from each treatment group to the control group (100%). One-way ANOVA tests, followed with Newman-Keuls multiple comparison tests, were subsequently performed to evaluate statistical differences between the various treatment groups. Statistical differences were considered significant at P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001). Data are represented as mean ± S.E.M. unless otherwise indicated.
3. Results
3.1. Co-administration of a selective Tachykinin NK1 receptor antagonist attenuates morphine withdrawal-mediated thermal hyperalgesia in rats
Similar to earlier data (King et al., 2005), we found that i.th. co-administration of the selective Tachykinin NK1 receptor antagonist, L-732,138 significantly attenuated morphine-withdrawal-mediated thermal hyperalgesia in rats. Baseline paw withdrawal latency in naïve animals (before drug intervention) was 24.4±1 sec (n=36; Fig. 1A). After baseline measurement, the rats were separated into treatment groups (n=6 animals in each group). Animals received twice daily i.p. morphine (or saline) injections with/without i.th. L-732,138 injections (refer to materials and methods). 96 h after the last drug treatment, paw withdrawal latencies were measured in each group (withdrawal hyperalgesia). Control animals treated with saline-saline exhibited mean paw withdrawal latencies similar to the mean baseline value ( 24.2±1 s; P>0.05 relative to baseline, n=6). Morphine withdrawal after sustained drug treatment (morphine-saline group) on the other hand, led to a marked decrease in mean paw withdrawal latency (11.3±1 s, ***P<0.001 relative to the saline-saline treated control, n=6). Interestingly however, rats receiving i.th. injections of the selective Tachykinin NK1 receptor antagonist (L-732,138 ) concurrent with i.p. morphine (morphine − L-732,138) exhibited paw withdrawal latencies ( 21.5±1 s; n=6) similar to the baseline (P>0.05) or saline-saline (P>0.05)-treated animal groups. This value was significantly different (# P< 0.01) from the mean paw withdrawal latency in the saline-morphine group. Withdrawal after sustained i.th. L-732,138 treatment did not cause significant difference in the paw withdrawal latencies of the animals, relative to the baseline and saline-saline groups (21.1±1s; n=6; P>0.05 vs. both baseline and saline-saline control) (Fig 1A).
Fig. 1. Selective Tachykinin NK1 receptor antagonists attenuate repeated opioid treatmentmediated A/thermal hyperalgesia and B/tactile allodynia in rats.
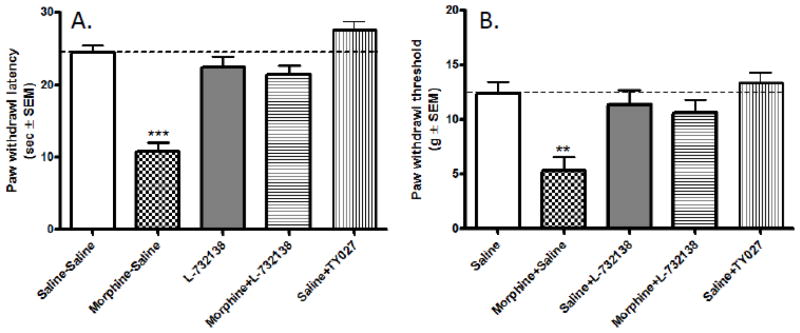
Rats received i.p. injections of saline (saline-saline control) or morphine (5mg/kg/injection; morphine-saline group) twice daily, for 6 days concurrent with i.th. injections of saline. The saline- L-732,138 and morphine− L-732,138 groups have received i.th. L-732,138 (20 μg/5 μl) injections concurrent with i.p. saline or morphine, respectively. In the Saline-TY027 group, the animals received repeated i.th. injections of a bivalent opioid agonist/Tachykinin NK1 receptor antagonist compound, TY027 (20 μg/5 μl/injection) twice daily for 6 days concurrent with i.p. saline injections. Thermal (A) and tactile (B) sensitivity of the animals was assessed prior to drug administration (naïve basal values - dotted lines) and 96 h after the last drug administration (bars).
3.2. Repeated administration of a bivalent opioid agonist/Tachykinin NK1 receptor antagonist compound (TY027) does not cause thermal hyperalgesia in rats
Baseline paw withdrawal latency in naïve animals (before drug intervention) was 24.4±1 sec (n=36; Fig. 1A). After baseline measurement, the rats were separated into treatment groups (n=6 animals in each group) and received i.th. injections (20 μg/5 μl, twice daily for 6 days) of TY027 ( a bivalent ligand having both opioid agonist activity and Tachykinin NK1 receptor antagonist activity) Interestingly, rats receiving sustained (6 days) i.th TY027 treatment and 96h drug withdrawal, exhibited paw withdrawal latencies ( 28.5±1 s; n=6) similar to the baseline. This value was significantly different (# P< 0.001) from the mean paw withdrawal latency in the saline-morphine group, indicating that withdrawal after i.th. TY027 treatment did not cause thermal hyperalgesia in rats (Fig. 1A).
3.3. Co-administration of a selective Tachykinin NK1 receptor antagonist (L-732,138 ) attenuates morphine withdrawal-mediated tactile allodynia in rats
The mean baseline paw withdrawal threshold value of naïve (before any drug intervention) rats used in the study was 13.7±1 g (n= 36) (Fig 1B). Sustained saline-saline treatment had no significant effect on the sensitivity of the animals toward mild tactile stimuli (12.5±2 g; P>0.05 relative to baseline, n=6; Fig. 1B). Drug withdrawal (96h) after sustained morphine treatment on the other hand, led to a significant decrease in the mean paw withdrawal threshold in the saline-morphine group (5 ± 2 g; **P<0.01 relative to the saline-saline treatment group and to the baseline values). Interestingly, rats receiving i.th. L-732,138 injections concurrent with the i.p. morphine injections (morphine−L-732,138 group) exhibited paw withdrawal thresholds (10.6±2 g, n=6) that are not significantly different from the mean baseline value (P>0.05) while being significantly different from the morphine-saline animal group (# P<0.05). Withdrawal after sustained L-732,138 treatment did not cause a significant difference in the paw withdrawal threshold of the animals (10.2±2 g; n=6; P>0.05 vs. baseline value and saline-saline control) (Fig 1B).
3.4. Repeated intrathecal administration of the bivalent opioid agonist/Tachykinin NK1 receptor antagonist does not cause tactile allodynia in rats
The mean baseline paw withdrawal threshold value of naïve (before any drug intervention) rats used in the study was 13.7±1 g (n= 36) (Fig 1B). Drug withdrawal (96h) after sustained (6 day) i.p. morphine administration led to a significant decrease in the mean paw withdrawal threshold in the saline-morphine group (5 ± 2 g; **P<0.01 relative to the saline-saline treatment group and baseline values). Interestingly however, rats receiving i.th. TY027 injections (20 μg/5 μl, twice daily for 6 days) after 96h drug withdrawal exhibited paw withdrawal thresholds (14.2±1 g, n=6) that were not significantly different from the mean baseline value (P>0.05) while being significantly different from the morphine-saline animal group (# P<0.001). This data indicates that withdrawal after repeated i.th. TY027 treatment did not cause tactile allodynia in the rats (Fig 1B).
3.5. L-732138 co-treatment normalizes microglia (OX-42) or astrocyte (GFAP) marker immunoreactivities in the lumbar spinal cord of morphine-withdrawn rats
After the behavioral assays, the animals were perfused and lumbar spinal cords were isolated from three animals in each treatment group. The intensity of immunofluorescence, measured as mean immunoflourescent pixels in control group for GFAP and OX-42 was 6210 ± 420 and 5400 ± 770, respectively (100%). Fluorescent immunohistochemistry in lumbar spinal cord slices shows that sustained morphine treatment caused a considerable increase in both OX-42- and GFAP-like immunoreactivities in the spinal cord of the rats (Figs. 2A and 2B). Normalized fluorescence intensities were 158 ± 8% of control for GFAP and 145 ± 10% of control for OX-42 (Fig. 2C;P<0.01, n=3, one-sample t-test)). L -732,138 co-administration completely attenuated sustained morphine-mediated augmentation of both OX-42 and GFAP-like immunoreactivities in the lumbar spinal cord of the animals (Fig. 2A and B). Quantification of immunohistochemical data (Fig 2C) showed that i.th. L -732,138 injections concurrent with the i.p. morphine injections inhibited sustained morphine-mediated spinal glial fibrillary acidic protein (114±7 relative to control; P<0.01 relative to morphine group, n=3, one-way ANOVA) and OX-42 immunoreactivity (110±9 relative to control; P<0.01 relative to morphine group, n=3, one-way ANOVA). These data indicate that sustained morphine-mediated spinal microglia and astrocyte activation can be inhibited by co-administration of a NK1 antagonist.
Fig. 2. Selective Tachykinin NK1 receptor antagonists attenuate repeated opioid treatment-mediated augmentation of A/microglia (OX-42) and B/astrocyte (GFAP) marker immunoreactivity in the lumbar spinal cord of opioid-withdrawn rats.

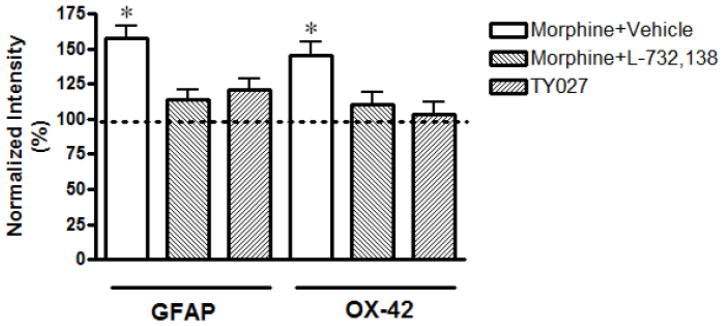
After drug treatments and behavioural tests, the rats were euthanized and their lumbar spinal cords were harvested. Serial spinal cord sections were mounted and incubated with (A) a mouse anti- OX-42 or (B) a mouse anti-glial fibrillary acidic protein (GFAP) antiserum followed by incubation with an Alexa Fluor 594-conjugated goat anti-mouse secondary antibody. Fluorescence images were digitally captured using a Nikon E800 fluorescence microscope. The acquired images were processed in Adobe PhotoShop. Insets show higher magnification images from similar dorsal horn areas of the sections. Quantification of the immunohistochemical data (C) was performed using the Image J software (NIH).
3.6. Microglia (OX-42) and astrocyte (GFAP) marker immunoreactivities are not elevated in the lumbar spinal cord of the bivalent opioid agonist/Tachykinin NK1 receptor antagonist-withdrawn rats
Fluorescent immunohistochemistry in lumbar spinal cord slices indicates that sustained morphine treatment caused a considerable increase in both OX-42- and GFAP-like immunoreactivities in the spinal cord (Fig 1A and 1B). Sustained i.th. TY027 injections on the other hand, did not augment microglia or astrocyte marker immunoreactivities in the lumbar dorsal horn of the rats. Quantification of immunohistochemical data (Fig 1B) indicates that the mean fluorescence intensity in this group was 121±8 relative to control for GFAP (*P<0.05 compared to morphine group) and 103±9 relative to control for OX-42 (*P<0.05 compared to morphine group, n=3, one-way ANOVA). These data indicate covalent linkage to a selective NK1 antagonist pharmacophore may counteract the spinal glial activation caused by the opioid agonist pharmacophore.
3.7. NK1 receptor antagonism attenuates opioid withdrawal-mediated augmentation of TNFα immunoreactivity in the spinal cord of rats
After behavioral tests, the animals were euthanized and lumbar spinal cords were isolated. TNFα immunoreactivity the homogenates was determined using a rat TNFα ELISA kit (Thermo Scientific). Mean TNFα concentration in the lumbar spinal cord of control animals was 94.4±2.6 pg/mL. Repeated morphine treatment and drug withdrawal significantly augmented spinal TNFα levels (115.4±10.4 pg/mL; P<0.05 relative to control). On the other hand, in animals receiving morphine+ L-732,138 or TY027 spinal TNFα levels were not significantly different from control (93.2±2.2 and 97.2±1.6 pg/mL, respectively, P>0.05 relative to control and P< 0.05 relative to the morphine-treatment group), indicating that NK1 antagonists normalize spinal pro-inflammatory mediator concentrations in opioid-withdrawn animals.
4. Discussion
Sustained administration of opioid analgesics increases the sensitivity of human patients (Ossipov et al., 2003; Simonnet and Rivat, 2003) and experimental animals (Mao et al., 1994; Vanderah et al., 2000; Gardell et al., 2002; Ossipov et al., 2003, King et al., 2005; Watkins et al., 2007; Mika et al., 2007) to mildly painful heat (thermal hyperalgesia) and normally innocuous mechanical (tactile allodynia) stimuli (opioid-induced hyperalgesia. Earlier data indicate that co-administration of opioid analgesics with a Tachykinin NK1 receptor antagonist attenuates sustained opioid agonist-mediated pain sensitization and antinociceptive tolerance (King et al., 2005). Our current data demonstrates that a/selective Tachykinin NK1 receptor antagonist co-administration prevents repeated morphine-mediated augmentation of microglia and astrocyte marker immunoreactivity in the spinal cord of rats; b/selective Tachykinin NK1 receptor antagonist co-administration prevents repeated morphine-mediated augmentation of spinal inflammatory mediator (TNFα) immunoreactivity and c/repeated treatment of rats with a bivalent opioid agonist/Tachykinin NK1 receptor antagonist compound does not augment spinal microglia or astrocyte marker or inflammatory mediator (TNFα) immunoreactivit in rats. Therefore, we suggest that spinal microglia/astrocyte Tachykinin NK1 receptors may provide a novel pharmacological target to prevent the spinal neuroinflammatory consequences of sustained opioid analgesic treatment.
Chronic pain seriously impairs the quality of life. Morphine, and other opioid agonists provide efficient relief from severe acute pain, but the use of opiates for the management of chronic and neuropathic pain is somewhat more problematic. First of all, opioid analgesics work less efficiently in neuropathic pain conditions and long-term drug administration further reduces their efficacy (Watkins et al., 2005, Watkins et al., 2007) and thus increased opiate doses are necessary to maintain the therapeutic effect. It was suggested earlier that a paradoxical pain sensitization contributes to the need for increased opiate doses during long-term pain management (apparent antinociceptive tolerance) (Mao et al., 1994; Vanderah et al., 2000; Ossipov et al., 2003, Mika et al., 2007).
Enhanced spinal excitatory pain neurotransmitter (such as Substance P) synthesis and/or release is one of the major adaptive changes associated with sustained opioid exposure (Ma et al., 2000; Ossipov et al., 2003, Powell et al., 2003; King et al., 2005, Vera-Portocarrero et al., 2007). Augmented spinal Substance P pain levels are thought to contribute to sustained opioid-mediated pain hypersensitivity and apparent antinociceptive tolerance (Ossipov et al., 2003, Powell et al., 2003; King et al., 2005, Vera-Portocarrero et al., 2007; Rivat et al., 2009). Indeed, co-administration of a Tachykinin NK1 receptor antagonist (SR140333) with morphine was found to attenuate opioid-jnduced hyperalgesia (King et al., 2005) and to prevent/reverse the development of antinociceptive tolerance (Powell et al., 2003). Furthermore, it was also found that genetic knockdown of the Substance P (NK1) receptor (King et al., 2005) or chemical ablation of the spinal SP-expressing peptidergic neurons (Rivat et al., 2009) attenuates the development of antinociceptive tolerance and paradoxical pain sensitization upon sustained opiate treatment in rodents. Based on these earlier findings, our group recently designed and synthesized promising novel bivalent antinociceptive drug candidates by covalently linking an opioid agonist pharmacophor (sterically constrained enkephalin analogues) with Substance P (SP) receptor (NK1) antagonists (Yamamoto et al., 2007, 2008). Many of the newly synthesized bivalent ligands (such as TY027; Yamamoto et al., 2007, 2008) were highly potent antinociceptive agents that produced no paradoxical pain sensitization and antinociceptive tolerance upon long-term administration in rats (Largent-Milnes et al., 2010b). The goal of the current project was to examine the cellular mechanisms contributing to attenuation of long-term opioid-mediated pain sensitization by Tachykinin NK1 receptor antagonists. It has been observed earlier that, similar to neuronal injury and inflammation (DeLeo and Yezierski, 2001; Raghavendra et al., 2003; Svennson et al., 2005), sustained morphine treatment activates CNS microglia and astrocytes (Raghavendra et al., 2004; Watkins et al., 2005, 2007; Mika et al., 2007) leading to pro-inflammatory modulator synthesis (Watkins et al., 2005; Raghavendra et al., 2004) in the spinal cord. Moreover, glial inhibitors (such as fluorocitrate, minocycline or pentoxyfylline) were found to attenuate sustained morphine-mediated pain sensitization and analgesic tolerance (Raghavendra et al., 2004; Watkins et al., 2005, 2007; Mika et al., 2007). Thus, co-administration of agents that attenuate glial activation during long-term opioid analgesic treatment is a promising novel way to attenuate paradoxical pain sensitization and antinociceptive tolerance in the clinical management of chronic pain. However, the currently available metabolic glial inhibitors are either toxic (fluorocitrate, (Willoughby et al., 2003)) or not selective for glia (also act as antibiotics (Graber et al., 1969) or as phosphodiesterase inhibitors (Yoshikawa et al., 1999)).
Microglia and astrocytes express numerous functionally active G protein-coupled receptors (Pocock and Kettenmann, 2007) offering novel phsarmacological targets to regulate glial activity. Thus, it was found earlier that CNS microglia and astrocytes express functional Substance P (Tachykinin NK1) receptors (Fiebich et al., 2000; Rassley et al., 2002, Marriott, 2004; Svensson et al., 2005; Hartel et al., 2009). Although SP is generally considered to be one of the chief pain neurotransmitters in the spinal cord (Allen et al., 1997), blocking SP activity by selective Tachykinin NK1 receptor antagonists has proven to be ineffective in blocking acute nociception in clinical trials (Hill, 2000; Herbert and Holzer, 2002). The reason for the failure of Tachykinin NK1 receptor antagonists to relieve acute pain is currently not clear.
Interestingly however, preclinical studies in animals indicate that Tachykinin NK1 receptor antagonists attenuate sensitized nociceptive responses due to inflammation and neuronal damage (Hill, 2000; Herbert and Holzer, 2002, Lee et al., 2007; Hamity et al., 2010). Furthermore, nerve injury and CNS infection upregulate glial Tachykinin NK1 receptor levels (Mantyh et al., 1989; Chauhan et al., 2008), indicating that the glial Tachykinin NK1 receptors may play a role in regulation of CNS neuroimmune responses (Marriott, 2004). Indeed, in vitro Tachykinin NK1 receptor activation in cultured microglia and in the spinal cord was shown to augment p38 MAP kinase phosphorylation (Fiebich et al., 2000; Svensson et al., 2005) and NF-κB-mediated pro-inflammatory modulator gene transcription (Fiebich et al., 2000; Marriott, 2004). Activation of astrocyte Tachykinin NK1 receptors on the other hand were shown to initiate intracellular Ca2+ transients (Hartel et al., 2009). Astrocyte Ca2+ waves are thought to regulate synaptic strength and contribute to long-term neuroplastic changes in the CNS (“tripartite synapses”) (Perea et al., 2009). Importantly, earlier data suggest that glial Tachykinin NK1 receptors may play a fundamental role in SP-mediated pain sensitization in the spinal cord, since antisense oligodeoxynucleotide knock-down of the microglial isoform of its crucial cellular effector (the β isoform of p38 mitogen-activated protein kinase (MAPK)) completely prevented intrathecal SP-mediated sensitization to chemical and mechanical stimuli in rats, while knockdown of the neuronal p38 MAPK α-isoform had no effect (Svensson et al., 2005 ).
In the present work we investigated the role of glial Tachykinin NK1 receptors in morphine withdrawal-mediated spinal microglia and astrocyte activation and spinal inflammatory mediator synthesis in rats. Our current results indicate that - in addition to acute activation of ascending neuronal pathways – sustained opioid-mediated spinal SP release may also have longer-lasting neuroinflammatory consequences by activating Tachykinin NK1 receptors on spinal microglia and/or astrocytes. By targeting spinal glial activation, co-administration of opioids with Tachykinin NK1 antagonists or bivalent opioid agonist/Tachykinin NK1 receptor antagonist may serve as novel, improved antinociceptive methods for long-term pain management.
Fig. 3. Selective Tachykinin NK1 receptor antagonists attenuate repeated opioid-mediated augmentation of spinal TNFα immunoreactivity in rats.
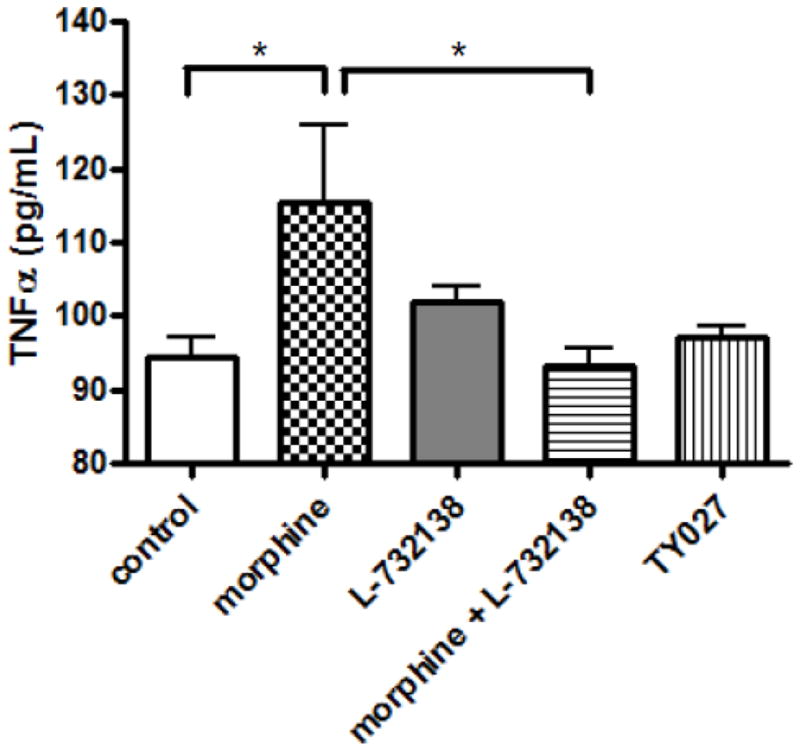
After drug treatment and behavioral test, the animals were euthanized and their spinal cords were isolated. Lumbar spinal cord segments were homogenized and TNFa immunoreactivty was measured using a Rat TNFα ELISA kit.
Acknowledgments
This work has been supported by grants (DA027786 and DA13449) from the National Institute of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen BJ, Rogers SD, Ghilardi JR, Menning PM, Kuskowski MA, Basbaum AI, Simone DA, Mantyh PW. Noxious cutaneous thermal stimuli induce a graded release of endogenous substance P in the spinal cord: imaging peptide action in vivo. J Neurosci. 1997;17:5921–5927. doi: 10.1523/JNEUROSCI.17-15-05921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan VS, Sterka DG, Gray DL, Bost KL, Marriott I. Neurogenic exacerbation of microglial and astrocyte responses to Neisseria meningitidis and Borrelia burgdorferi. J Immunol. 2008;180:8241–8249. doi: 10.4049/jimmunol.180.12.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Schleicher S, Butcher RD, Craig A, Lieb K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa. Br J Immunol. 2000;165:5606–5611. doi: 10.4049/jimmunol.165.10.5606. [DOI] [PubMed] [Google Scholar]
- Freye E, Latasch L. Development of opioid tolerance - Molecular mechanisms and clinical consequences. Anasthesiol Intensivmed Notfallmed Schmerzther. 2003;38:14–26. doi: 10.1055/s-2003-36558. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber CD, Jervey LP, Martin F, Boltjes BH. In vitro and in vivo sensitivity of staphylococci and selected bacteria to minocycline, tetracycline and doxycycline. J S C Med Assoc. 1969;65:197–200. [PubMed] [Google Scholar]
- Hamity MV, White SR, Hammond DL. Effects of neurokinin-1 receptor agonism and antagonism in the rostral ventromedial medulla of rats with acute or persistent inflammatory nociception. Neuroscience. 2010;165:902–913. doi: 10.1016/j.neuroscience.2009.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Härtel K, Schnell C, Hülsmann S. Astrocytic calcium signals induced by neuromodulators via functional metabotropic receptors in the ventral respiratory group of neonatal mice. Glia. 2009;57:815–827. doi: 10.1002/glia.20808. [DOI] [PubMed] [Google Scholar]
- Herbert MK, Holzer P. Why are substance P (NK1)-receptor antagonists ineffective in pain treatment? Anaesthesist. 2002;51:308–319. doi: 10.1007/s00101-002-0296-7. [DOI] [PubMed] [Google Scholar]
- Hill R. NK1 (substance P) receptor antagonists--why are they not analgesic in humans? Trends Pharmacol Sci. 2000;21:244–246. doi: 10.1016/s0165-6147(00)01502-9. [DOI] [PubMed] [Google Scholar]
- King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP, Vanderah TW, Hunt SP, Hruby VJ, Lai J, Porreca F. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005;116:276–88. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largent-Milnes TM. Doctoral Thesis. The University of Arizona; 2010a. Neurokinin 1 receptors and their role in opioid-induced hyperalgesia, antinociceptive tolerance and reward. [Google Scholar]
- Largent-Milnes TM, Yamamoto Y, Nair P, Moulton JW, Hruby VJ, Lai J, Porreca F, Vanderah TW. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol. 2010b;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Kim JH. Involvement of substance P and calcitonin gene-related peptide in development and maintenance of neuropathic pain from spinal nerve injury model of rat. Neurosci Res. 2007;58:245–249. doi: 10.1016/j.neures.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Kar S, Quirion R. Morphine treatment induced Calcitonin Gene-related Peptide and Substance P increases in cultured Dorsal Root Ganglion neurons. Neuroscience. 2000;99:529–539. doi: 10.1016/s0306-4522(00)00226-8. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Johnson DJ, Boehmer CG, Catton MD, Vinters HV, Maggio JE, Too HP, Vigna SR. Substance P receptor binding sites are expressed by glia in vivo after neuronal injury. Proc Natl Acad Sci U S A. 1989;86:5193–5197. doi: 10.1073/pnas.86.13.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Thermal hyperalgesia is associated with the development of morphine tolerance in rats: Roles of excitatory amino acid receptors and Protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott I. The role of tachykinin in central nervous system inflammatory responses. Front Biosci. 2004;9:2153–2165. doi: 10.2741/1377. [DOI] [PubMed] [Google Scholar]
- Mika J, Osikowicz M, Makuch W, Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol. 2007;560:142–149. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Vanderah TW, Porreca F. Induction of pain facilitation by sustained opioid exposure: Relationship to opioid antinociceptive tolerance. Life Sci. 2003;73:783–800. doi: 10.1016/s0024-3205(03)00410-7. [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci. 2003;18:1572–1583. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacol. 2004;29:327–334. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- Rasley A, Bost KL, Olson JK, Miller SD, Marriott I. Expression of functional NK-1 receptors in murine microglia. Glia. 2002;37:258–267. doi: 10.1002/glia.10034. [DOI] [PubMed] [Google Scholar]
- Rivat C, Vera-Portocarrero LP, Ibrahim MM, Mata HP, Stagg NJ, De Felice M, Porreca F, Malan TP. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci. 2009;29:727–37. doi: 10.1111/j.1460-9568.2009.06616.x. [DOI] [PubMed] [Google Scholar]
- Simonnet G, Rivat C. Opioid-induced hyperalgesia: abnormal or normal pain? Neuroreport. 2003;14:1–7. doi: 10.1097/00001756-200301200-00001. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL. Spinal p38beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J Neurochem. 2005;92:1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, Porreca F. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007;129:35–45. doi: 10.1016/j.pain.2006.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson HR, Johnston JN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends in Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–146. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby JO, Mackenzie L, Broberg M, Thoren AE, Medvedev A, Sims NR, Nilsson M. Fluorocitrate-mediated astroglial dysfunction causes seizures. J Neurosci Res. 2003;74:160–166. doi: 10.1002/jnr.10743. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nair P, Davis P, Ma SW, Navratilova E, Moye S, Tumati S, Lai J, Vanderah TW, Yamamura HI, Porreca F, Hruby VJ. Design, synthesis, and biological evaluation of novel bifunctional C-terminal-modified peptides for delta/mu opioid receptor agonists and neurokinin-1 receptor antagonists. J Med Chem. 2007;50:2779–2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma SW, Navratilova E, Moye S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. The importance of micelle-bound states for the bioactivities of bifunctional peptide derivatives for delta/mu opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2008;51:6334–6347. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M, Suzumura A, Tamaru T, Takayanagi T, Sawada M. Effects of phosphodiesterase inhibitors on cytokine production by microglia. Mult Scler. 1999;5:126–133. doi: 10.1177/135245859900500210. [DOI] [PubMed] [Google Scholar]