

Abstract
Background
A leptosomic body type is tall and thin with long hands. Marfanoid features may be familial in nature or pathological, as occurs in congenital contractual arachnodactyly (Beal’s syndrome) and Shprintzen-Goldberg syndrome mimicking some of the changes of Marfan syndrome, although not accompanied by luxation of lens and dissecting aneurysm of aorta.
Methods
In this article we collected eight patients who were consistent with the diagnosis of Marfan syndrome via phenotypic and genotypic characterization.
Results
Our patients manifested a constellation of variable presentations of musculo-skeletal abnormalities ranging from developmental dysplasia of the hip, protrusio acetabuli, leg length inequality, patellar instability, scoliosis, to early onset osteoarthritis. Each abnormality has been treated accordingly.
Conclusion
This is the first paper which includes the diagnosis and the management of the associated musculo-skeletal abnormalities in patients with Marfan syndrome, stressing that patients with Marfan syndrome are exhibiting great variability in the natural history and the severity of musculo-skeletal abnormalities.
Keywords: Marfan syndrome, musculo-skeletal abnormalities, phenotype, genotype, surgery
Introduction
Patients with Marfan syndrome are a clinically diverse group. They can have any combination of dolichostenomelia, arachnodactyly, pectus deformities of the chest, mitral or aortic regurgitation, ectopia lentis, and mild joint laxity. Other evidence of a generalized connective tissue disorder may be present such as scoliosis and skin striae. Patients with Marfan syndrome are typically tall, usually attaining a height greater than 6 feet in adult life, and have disproportionately long, thin limbs. The distal bones of the limbs exhibit the excess length most strikingly, resulting in long, slender hands and feet with spider-like digits, a condition known as arachnodactyly.1,2 Musculoskeletal abnormalities such as pectus excavatum are caused by excessive longitudinal growth of the ribs. Pectus carinatum is also frequently seen in patients with Marfan syndrome. Joint laxity is another hallmark of the disease. Dislocation of the hip, either developmental or presenting later, and other joints can occur. The vertebral column is significantly affected in patients with Marfan syndrome.3 Congenital hip dislocation in Marfan syndrome (infantile type) and protrusio acetabuli can be seen radiographically and may manifest as hip pain and stiffness.4–6 In our series of patients, protrusio acetabuli, early-onset osteoarthritis, patellar instability, limb length inequality, developmental dysplasia of the hip and scoliosis were the most prominent skeletal abnormalities identified.
Methods
The study protocol was approved by the Medical University of Vienna (Ethics Committee, EK Nr. 921/2009), and informed consents were obtained from the patients’ guardians.
Our patients were of different ethnic origins. The patients’ records were reviewed in the Department of Genetic Bone Disorders at the Orthopaedic Hospital of Speising in Vienna, Austria. Extensive search for specific phenotypic features, natural history, and skeletal surveys were performed to prepare this paper, which is illustrative of the spectrum of musculoskeletal abnormalities in Marfan syndrome. A patient with classic florid Marfan syndrome is easily recognized as an unusually tall, lanky individual with arachnodactyly, disproportionate long limbs, chest wall deformity, severe myopia, and a loud cardiac murmur. The ratio of the upper body segment (measures from the top of the symphysis pubis to the top of the head, or by subtracting the measurement of the lower body segment from the total height) to the lower body segment (measured from the sole to the top of the symphysis pubis) is abnormally low (the upper body-lower body ratio is 0.93 in the normal adult population). In addition, the arm span exceeds the patient’s total height. Skeletal abnormalities such as pectus excavatum are a common manifestation and are caused by excessive longitudinal growth of the ribs. The anteroposterior diameter of the thoracic cage is reduced. Significant joint laxity is another hallmark of the disease.
The clinical diagnosis of Marfan syndrome was based on Ghent diagnostic nosology devised in 1996, as a revision of the earlier Berlin criteria of 1988. In this nosology, clinical features are assessed as part of the seven body system to determine whether that system provides a major criterion, or simply multiple system involvement (Table 1). All our Marfan patients were genetically tested for mutation in the fibrillin I gene on chromosome 15. Mutations have been encountered in all our patients.
Table 1.
Ghent diagnostic nosology as applied to our patients.
| System | Major criteria | Involvement |
|---|---|---|
| Pectus carinatum | Pectus carinatum in 5 patients | Pectus excavatum in 3 patients |
| Upper to lower segment ratio (ULSR) < 0.86 | Upper to lower segment ratio (ULSR) < 0.86; in all patients | All manifested ligamentous hyperlaxity |
| Wrist and thumb sign | Wrist and thumb sign; in all patients | High palate and dental crowding in 5 patients |
| Scoliosis > 20° | Scoliosis > 20° in 2 patients | All manifested the characteristic facial features |
| Reduced elbow extension < 170° | Reduced elbow extension < 170°; All patients | |
| Pes planus | All patients | |
| Protrusio acetabuli | Protrusio acetabuli in 4 patients | |
| Lens dislocation | All patients | |
| Cardiovascular | Five patients showed dilatation of the aortic root and 1 patient operated for dissecting aortic aneurysm | 3 patients with mitral valve prolapse |
| Pulmonary | None | |
| Genetic findings | All showed fibrillin 1 mutation |
Developmental dysplasia of the hip
The estimated incidence of a dislocated hip (DDH) at birth in the general population is 0.65 to 1.5 per 1000 live births. The cause of DDH is multifactorial including prenatal and perinatal positioning, ligamentous hyperlaxity, and several forms of skeletal dysplasias.4
Patients with Marfan syndrome may present developmental dysplasia of the hip as newborns or infants. We diagnosed one female patient presented with infantile type or congenital Marfan syndrome. Infantile scoliosis associated with bilateral hip dislocation were the major musculoskeletal abnormalities. Interestingly, the child was a product of a Marfan syndrome father (autosomal dominant). Pavlik harness was unsuccessful in obtaining reduction of the hip, and closed reduction under anaesthesia was planned for (Fig. 1).
Figure 1.

Dislocation of the hip and infantile scoliosis in a 7-month-old girl manifesting the full clinical criteria of Marfan syndrome, next to her is her father where arachnodactyly can be noted to confirm the autosomal dominant pattern of inheritance.
Protrusio-acetabuli
Intrapelvic protrusio acetabuli, is a deformity of the hip joint in which the medial wall of the acetabulum invades the pelvic cavity with associated medial displacement of the femoral head.5 Three patients manifested protrusio-acetabuli (a condition which is characterized clinically by hip joint stiffness and pain and radiologically by collapse of the acetabular teardrop) (Fig. 2). Prolonged protrusio acetabuli may result in secondary osteoarthritic changes in the hip joint. Radiographic criteria for protrusion acetabuli include an abnormally positioned acetabular line, a center-edge angle of Wiberg of >40°, and the crossing of the teardrop by the ilioischial line. In a skeletally immature patient with Marfan syndrome in whom the triradiate physis of the acetabulum is still open, closure of the triradiate physis can interrupt and decrease the progression of the deformity. In older patients, valgus intertrochanteric osteotomy and eventually total hip arthroplasty are the only methods available for correction of the protrusio acetabuli (Fig. 3).
Figure 2.

AP radiograph of the hips in a 33-year-old patient with Marfan syndrome showed protrusio acetabuli in which there is abnormally positioned acetabulae associated with severe osteoarthritis.
Figure 3.

AP radiograph of the hip shows total hip arthroplasty for correction of the protrusio-acetabuli after valgus intertrochanteric osteotomy.
Leg length inequality
Leg length inequality in children is a frequent parental concern or physical finding noted by the orthopaedist.8 Asymptomatic leg length inequality is relatively common in the healthy paediatric and adult population. Leg length inequality has been recognized in one male patient. The limb inequality in this patient was secondary to a unilateral protrusio acetabuli.
A 15-year-old male patient presented with a 41 mm limb length discrepancy. Therefore, based on Canale, we performed permanent epiphysiodesis with drilling (Fig. 4).
Figure 4.

A 15-year-old male patient presented with a limb length discrepancy of 41 mm. A permanent epiphysiodesis with drilling has been applied based on Canale.
Patellar instability
Recurrent Patellar Instability (RPI) is defined as more than one episode of dislocation of the patella documented by an observer or clearly described by the patient. The dislocations are almost always to the lateral side of the femur. The investigators found that repair of the MPFL restored stability and that repair of secondary retinaculum structures, the patellotibial and patellomeniscal complex, provided no increase in stability. Sky line view of a 10-year old boy with Marfan syndrome showed bilateral patellar dysplasia and instability (lateralization of the patellae), defective ossification of the patellae, and trochlear dysplasia. Note pathological features of loss of the articular cartilage and alterations in the shape of the articular surface (Fig. 5).
Figure 5.
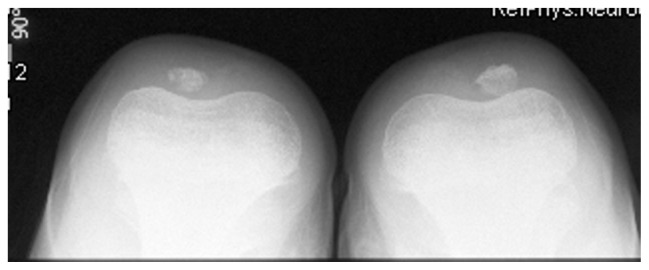
Sky line view of a 10-year old boy with Marfan syndrome showed bilateral patellar dysplasia and instability (lateralization of the patellae), defective ossification of the patellae and trochlear dysplasia. Note pathological features of loss of the articular cartilage and alterations in the shape of the articular surface.
Scoliosis
Scoliosis affects around 60% of Marfan patients and there may be rapid progression during growth spurts, leading to marked deformity, pain, and restricted ventilatory deficit. In adult patients, back pain caused by scoliosis is three times more frequent than in the general population.3 Two patients out of eight were identified who developed scoliosis of more than 45°.
A 19-year-old female patient was first seen in the outpatient department in 2006, presenting with a scoliosis of the thoracolumbar spine. She reported of an augmenting side bolster on her loins. X-rays showed a scoliosis of 60° Cobb left-convex with an apex at the first/second lumbar vertebral body. There were no signs of neurologic symptoms.
Reformatted CT scan showed the progressive deformation of the thoraco-lumbar vertebrae in a patient with Marfan syndrome, there is loss of the normal vertebral concavity associated with progressive antero-lateral growth reduction with exaggeration of vertebral flattening associated with the development of marginal osteophytes (arrow) T12, L1-2 (bony outgrowths of the vertebral column) which are suggestive of spinal arthritis secondary to progressive degeneration with subsequent development of endplate abnormality and or destruction. Ligamentous laxity is also a cause of the precocious axial and extra-axial osteoarthritis in Marfan syndrome (Fig. 6). Because of an insufficient bending in the preoperative radiographs, the operative procedure was planned in two steps. First, a ventral release was done at the level L1/L2, L2/L3 and L3/4. Seven days later, the dorsal fusion with instrumentation of L5, L4, L3, Th 8, Th 9 with screws in every pedicel and a single screw on L1 (convexity) and Th 12 (concavity) was performed, and fixation of the bar in the right position was done. After neurologic control in anesthesia, the fusion was prepared by inserting autologous bone and osteoinductive bone substitute (Fig. 7).
Figure 6.

Reformatted CT scan showed the progressive deformation of the thoraco-lumbar vertebrae in a patient with Marfan syndrome, there is loss of the normal vertebral concavity associated with progressive antero-lateral growth reduction with exaggerated vertebral flattening associated with the development of marginal deformity (arrow) T12, L1-2 (bony outgrowths of the vertebral column) which are suggestive of spinal arthritis secondary to progressive degeneration with subsequent development of end-plate abnormality and or destruction.
Figure 7.
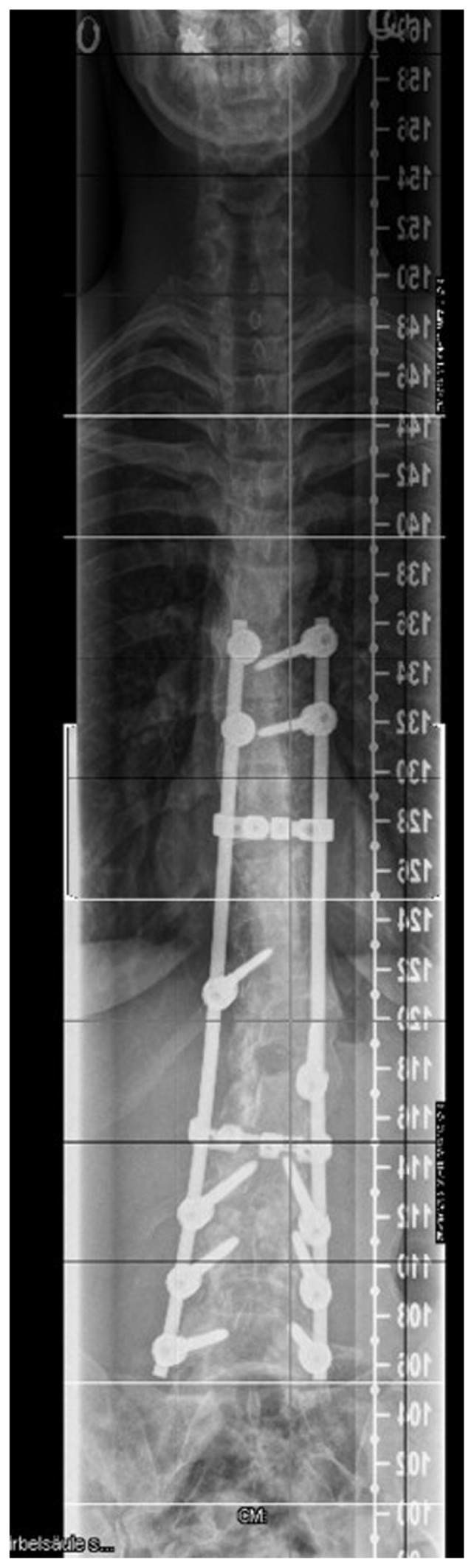
The operative procedure was planned in two steps, because of an insufficient bending in the preoperative radiographs. First, a ventral release was done at the level L1/L2, L2/L3 and L3/4. 7 days later, the dorsal fusion with instrumentation of L5, L4, L3, Th 8, Th 9 with screws in every pedicle and a single screw on L1 (convexity) a Th 12 (concavity) was performed, and fixated on the bar in the right position. After neurologic control in anesthesia the fusion was prepared by inserting autologous bone and osteoinductive bone substitute.
Osteoarthritis
Osteoarthritis (OA) or degenerative joint disease (DJD) is more common than all the other types of arthritis combined. It is well-established that injury to a joint increases the chances that the joint will develop OA over time. Precipitating causes include sudden impact or trauma, overuse or repetitive motion injuries, biomechanical abnormalities (congenital or acquired), ligamentous injury, joint hypermobility, obesity, intra-articular or systemic corticosteroids, avascular necrosis, and hereditary factors. OA, though the accepted term used to describe DJD, is misleading as it primarily relates to cartilage, not bone, and involves degeneration, not inflammation. Ligamentous damage or weakness is one cause of joint degeneration. Joint subluxations, dysplasia, and incongruity prevent the normal distribution of weight and stresses on the articular surfaces of the joint leading to cartilage injury and joint degeneration.9,10 AP radiograph of the knee showed severe OA matching the grade of II–III of Kellgren-Lawrence grading scale.11 The flattened and dysplastic epiphyses associated with marked narrowing of the joint spaces, sclerosis, and subsequent deformity of the bony contour were noted. This resulted in the patient using crutches to walk. The knee was unstable medial and lateral, patella was sub-luxated to the lateral and medial side, atrophy of the quadriceps muscle was evident, and the MRI indicated an infarction area in the distal femur (Fig. 8). Recently, a total knee prosthesis was implanted (LCCK), healing occurred without complications. Mobilization afterwards was very slow, the patient used a wheelchair because of instability in ankle-joints, arthritis and instability in the left knee was evident (Fig. 9).
Figure 8.
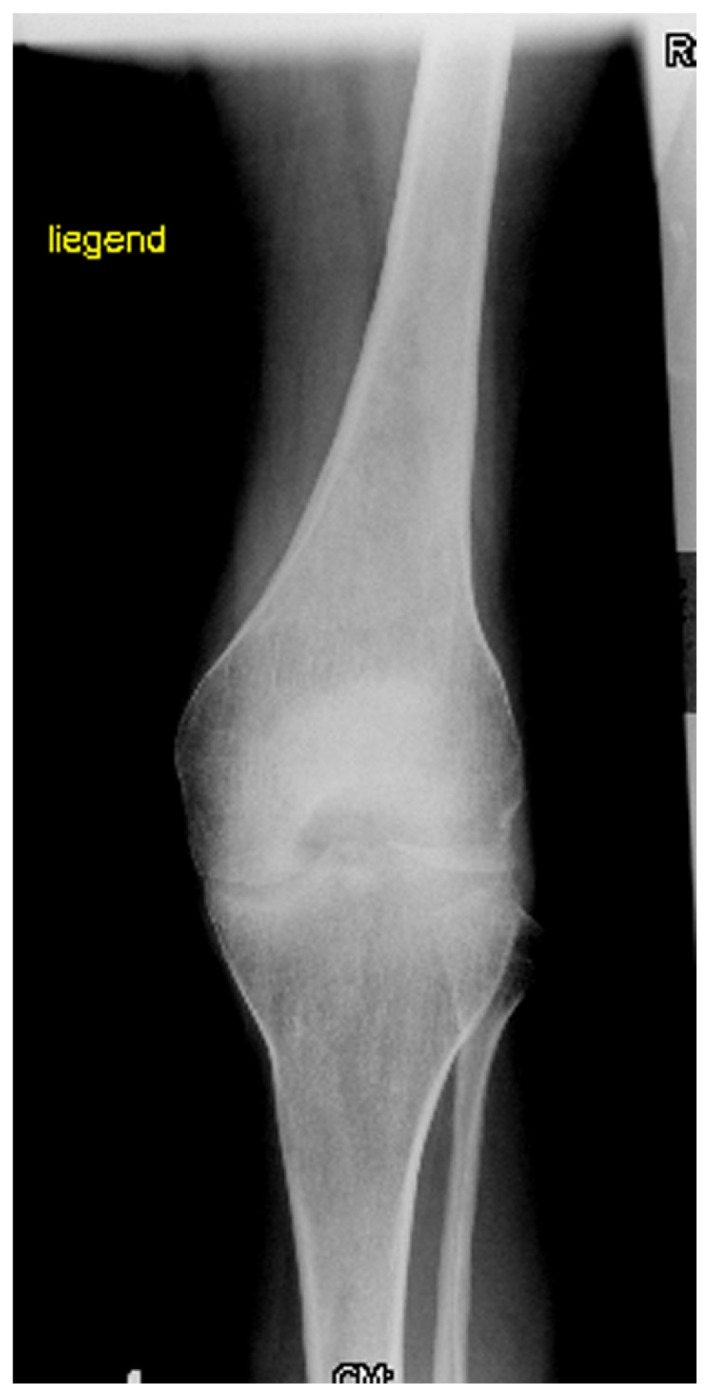
AP radiograph of the knee showed severe osteoarthritis matching the grade of II–III of Kellgren-Lawrence grading scale. Note the flattened and dysplastic epiphyses associated with a marked narrowing of the joint spaces, sclerosis and subsequent deformity of the bony contour, the patient started to walk with crutches. The knee was unstable medial and lateral +++, patella was sub-luxated to the lateral and medial side, atrophy of the quadriceps muscle was evident, the MRI indicated an infarction-area in the distal femur.
Figure 9.

Recently a total knee prosthesis was implanted (LCCK), healing occurred without complications. Mobilization afterwards was very slow, the patient used a wheelchair because of instability in the anklejoints, arthritis and instability in the left knee was evident.
Genetics
Marfan syndrome is caused by a defective gene FBN1, located on the long arm of chromosome 15. This gene encodes for fibrillin-1, a large glycoprotein closely associated with elastin. In addition to their presence in the aortic media and suspensory ligaments of the lens, fibrillin microfibrils are found in skin, tendon, cartilage, and periosteum. In contradistinction, the much rarer Beal’s syndrome is caused by the defective gene FBN2, located on chromosome 5 that encodes for the glycoprotein fibrillin-2.
Discussion
Marfan syndrome is a complex disease characterized by abnormally long, thin extremities and associated with eye and cardiovascular anomalies. Autosomal dominant inheritance with high penetrance and wide expressive variability is an additional criterion of the disease. The main clinical features are dolichocephaly with frontal bossing, long narrow face with prominent mandible, high arched palate, and narrow thorax with pigeon or funnel chest. The limbs are characterized of being long (dolichostenomelia) and long thin fingers (like the legs of a spider). Flatfoot deformity and skewfoot deformity, with hind foot valgus and forefoot adductus or varus, are not uncommon features of the disease. The skin looks dry and wrinkled with minimal subcutaneous fat. Ophthalmological abnormalities of dislocated lens, iridodonesis, spherophakia, and severe myopia are well documented abnormalities in connection with Marfan syndrome. Congenital anomalies of the cardiovascular system, such as septal defects, valvular anomalies, and vascular hypoplasia as well as the acquired pathology of dilatation of the ascending aorta and dissecting aortic aneurysm, are serious complications of the disease.1,7,12 The pathological mechanism by which mutation in FBN1 fibrillin-1 encoding gene predisposes to the constellation of the musculo-skeletal abnormalities encountered in Marfan Patients is mostly based on the presence of fibrillin-1 mutation gene in cartilage, periosteum, and skin. Fibrillin-1 gene in the cartilage, results in various abnormalities, namely developmental dysplasia of the hip and protrusio acetbulae and other weight bearing zones abnormalities.6 In addition, the presence of a mutated gene in the aortic media and in the suspensory ligaments is behind the pathological abnormalities of the cardiovascular and the eyes abnormalities noted in Marfan patients. Mutations in fibrillin-1 protein can also be encountered in other syndromic entities such as isolated ectopia lentis, Weil-Marchesani syndrome, and Shprintzen-Goldberg syndrome.13,14
Protrusio acetabuli is clinically characterized by hip/joint stiffness and pain, and radiologically by collapse of the acetabular teardrop. Steel described surgical closure of the triradiate cartilage in skeletally immature patients who were symptomatic or in whom increasing acetabular deeping was demonstrated radiologically.15 In his report on 19 hips followed to skeletal maturity, radiographic indices normalized in 12 cases, improved in 4, and were unchanged in 3. All hips were asymptomatic. He recommended surgical closure of the triradiate cartilage for patients between 8 and 10 years of age. Höhle discovered protrusio acetabuli in two of his patients who had arachnodactyly, keel breast, ectopia lentis, and a familial incidence of protrusion acetabuli.16 Nevertheless, he stated that the aetiology and pathogenesis of idiopathic protrusion acetabuli is not yet explained in all aspects. Later on, Steel reported seven cases of protrusion acetabuli, five of which were in patients who had all of the clinical manifestations of Marfan syndrome. Older patients had symptomatic relief but little improvement in the radiographic appearance of their hips.12 Others have reported that the occurrence of protrusio acetabuli in patients with Marfan syndrome is correlated to disturbed bone mineral density and that it is as high as 31%. Prolonged protrusio acetabuli may result in secondary osteoarthritic changes in the hip joint.16,17
The generalized joint laxity that characterizes patients with Marfan syndrome may manifest as severe pes planovalgus or joint dislocations, especially of the patello-femoral joint.7,9,18
In patients with Marfan syndrome, ligamentous hyperlaxity is a predominating feature, in which the primary medial soft tissue stabilizer of the patella is the medial patello-femoral ligament (MPFL). This is an extra capsular, ligamentous thickening of varying width within the medial retinaculum. In one study, release of this ligament resulted in a 50% increase in lateral displacement of the patella with loading.
Scoliosis affects around 60% of Marfan patients and there may be rapid progression during growth spurts, leading to marked deformity, pain, and restricted ventilatory deficit. In adults, back pain caused by scoliosis is three times more frequent than in the general population. Radiographs typically show tall vertebral bodies with elongated transverse processes. The position of the sacrum is low in relation to the iliac crests. The spinal canal may appear widened in the lumbar region, with concavity of the posterior borders of the vertebral bodies.3 Reformatted CT scan in two of our scoliotic patients showed loss of the normal vertebral concavity associated with progressive antero-lateral growth reduction with exaggerated vertebral flattening associated with the development of marginal osteophytes (bony outgrowths of the vertebral column) suggestive of spinal arthritis secondary to progressive degeneration with subsequent development of endplate abnormality and or destruction. Ligamentous laxity is also a cause of the precocious spinal OA in Marfan syndrome.
A lack of understanding about the development of OA has resulted in a broad array of symptom-based treatment options such as analgesics, anti- inflammatory, braces and wraps, physical therapy and exercise, chiropractic, viscosupplementation, corticosteroid injections, and surgery. While advances have been made in joint replacement, cartilage repair, cartilage replacement, and spinal procedures, treatments to limit or even reverse articular cartilage breakdown have been lacking. Ligament injury, excess laxity, joint hypermobility, and clinical instability are known to be major causes of OA. Therefore, any treatment which can address restoration of ligament function would help reduce the incidence, pain, and dysfunction of OA should be based on etiological understanding of the underlying pathology.10,11,19
Average life expectancy of Marfan patients is halved in comparison to the general population. 95% of deaths are due to a cardiovascular cause. Shores et al studied the effect of beta-adrenergic blockers and concluded that this slowed the rate of aortic dilatation and reduced the development of complications from aortic rupture in some patients with Marfan syndrome.20 There have been approximately 10 case reports of cerebral aneurysms in Marfan syndrome, but the association is doubted by some.19 The Committee on Genetics of the American Academy of Pediatrics provides guidelines for management during childhood.21,22
The differential diagnosis of Marfan syndrome from other disorders of Marfanoid habitus is mandatory. Congenital contractual arachnodactyly (Beal’s syndrome), and Shprintzen-Goldberg syndrome mimic some of the changes of Marfan syndrome, without being accompanied by luxation of the lens, funnel chest, and dissecting aneurysm of aorta.23,24
Homocystinuria, Stickler syndrome, marfanoid hypermobility syndrome, familial or isolated mitral valve prolapse syndrome, familial or isolated annuloaortic ectasia, familial idiopathic dilatation of the aorta with dissection without Marfan syndrome; Lujan-Fryns syndrome (X-linked mental retardation with Marfanoid habitus).25 De Becker et al described that the expressions of Marfan syndrome can vary and might overlap with Ehlers-Danlos (kyphoscoliotic type).26
Conclusion
In patients with Marfanoid habitus, screening for Marfan syndrome should be performed according to the Ghent criteria. Although the defective gene for Marfan syndrome has been identified, the diagnosis remains a clinical one, based on the fulfillment of diagnostic criteria. Clinicians, particularly primary care physicians and those working in scoliosis clinics must be aware of the possibility of this condition because the presenting anomaly is most commonly either scoliosis or a heart murmur. The physician should seek a family history of the disorder or its manifestations, especially tall stature, skeletal abnormalities, ligament hyperlaxity, poor vision, cardiac anomalies, and sudden or premature death.
Acknowledgement
We wish to thank Dr. Franco Laccone Department für Medizinische Genetik. Medizinische Universität Wien, for his help in performing the genetic tests.
Footnotes
Author Contributions
Drafted the manuscript and analysed the data: AAK. Participated in coordination of the study: EZ, KK. Participated in its design and coordination: RG, SS, FG. All authors read and approved the final manuscript.
Competing Interests
EZ has received payment for lectures. Other authors disclose no competing interests.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Funding
Author(s) disclose no funding sources.
References
- 1.Marfan AB. Un cas de déformation des quatre membres plus prononcé aux extrémités caractérisé par l’allongement des os avec un certain degré d’amainssissement. Bull Men Soc Med Hop (Paris) 1886;13:220. [Google Scholar]
- 2.Faivre L, Collod-Beroud G, Adès L, et al. The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet. 2012;81(5):433–42. doi: 10.1111/j.1399-0004.2011.01703.. [DOI] [PubMed] [Google Scholar]
- 3.Daeubler BF, Carrel T, Kujawski T, et al. Alterations of the thoracic spine in Marfan’s syndrome. AJR Am J Roentgenol. 2006;186(5):1246–51. doi: 10.2214/AJR.05.0071. [DOI] [PubMed] [Google Scholar]
- 4.Wiberg G. Studies on dysplastic acetabula and congenital subluxation of the hip joint. With special reference to the complication of osteoarthritis. Acta Chir Scand. 1939;83(Suppl 58):5–135. [Google Scholar]
- 5.Van de Velde S, Fillman R, Yandow S. Protrusio acetabuli in Marfan syndrome. History, diagnosis, and treatment. J Bone Joint Surg Am. 2006;88(3):639–49. doi: 10.2106/JBJS.E.00567. [DOI] [PubMed] [Google Scholar]
- 6.Sponseller PD, Tomek IM, Pyertiz RE. Developmental dysplasia of the hip in Marfan syndrome. J Pediatr Orthop B. 1997;6(4):225–9. doi: 10.1097/01202412-199710000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Joseph KN, Kane HA, Milner RS, et al. Orthopaedic aspects of the Marfan phenotype. Clin Orthop Relat Res. 1992;(277):251–61. [PubMed] [Google Scholar]
- 8.Soukka A, Alaranta H, Tallroth K, Heliovaara M. Leg length inequality in people at working age. The association between mild inequality and low-back pain is questionable. Spine (Phila Pa 1976) 1991;16(4):429–31. doi: 10.1097/00007632-199104000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Warren LF, Marshall JL. The supporting structures and layers on the medial side of the knee. An anatomical analysis. J Bone Joint Surg Am. 1979;61(1):56–62. [PubMed] [Google Scholar]
- 10.Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: New insights. Part 1: The disease and its risk factors. Ann Intern Med. 2000;133(8):635–46. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 11.Kellgren JH, Lawrence JS. Radiological assessment of osteoarthritis. Ann Rheum Dis. 1957;16(4):494–502. doi: 10.1136/ard.16.4.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pyeritz RE. The Marfan syndrome. In: Royce PM, Steinman B, editors. Connective Tissue and Its Heritable Disorders. New York, NY: Wiley-Liss; 1993. pp. 437–68. [Google Scholar]
- 13.Robinson PN, Godfrey M. The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet. 2000;37:9–25. doi: 10.1136/jmg.37.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson PN, Arteaga-Solis E, Baldock C, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43(10):769–87. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steel HH. Protrusio acetabuli: Its occurrence in the completely expressed Marfan syndrome and its musculoskeletal components and a procedure to arrest the course of protrusion in the growing pelvis. J Pediatr Orthop. 1996;16:704. doi: 10.1097/00004694-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Höhle B. Familial occurrence of protrusion acetabuli. Beitr Orthop Traumatol. 1978;25(5):261–5. German. [PubMed] [Google Scholar]
- 17.Do T, Giampierto PF, Burks SW, et al. The incidence of protrusio acetabuli in Marfan’s syndrome and its relationship to bone mineral density. J Pediatr Orthop. 2000;20(6):718–21. doi: 10.1097/00004694-200011000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Joseph KN, Kane HA, Milner RS, Steg NL, Williamson MB, Jr, Bowen JR. Orthopedic aspects of the Marfan phenotype. Clin Orthop Relat Res. 1992;(277):251–61. [PubMed] [Google Scholar]
- 19.Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: New insights. Part 1: The disease and its risk factors. Ann Intern Med. 2000;133(8):635–46. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 20.Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. New Engl J Med. 1994;330(19):1335–41. doi: 10.1056/NEJM199405123301902. [DOI] [PubMed] [Google Scholar]
- 21.van den Berg JS, Limburg M, Hennekam RC. Is Marfan syndrome associated with symptomatic intracranial aneurysms? Stroke. 1996;27:10–2. doi: 10.1161/01.str.27.1.10. [DOI] [PubMed] [Google Scholar]
- 22.Desposito F, Cho S, Frias JL, et al. American Academy of Pediatrics. Health supervision for children with Marfan syndrome. Committee on genetics. Pediatrics. 1996;98:978–82. [PubMed] [Google Scholar]
- 23.Beals RK, Hecht F. Congenital contractural arachnodactyly: a heritable disorder of connective tissue. J Bone Joint Surg Am. 1971;53(5):987–93. [PubMed] [Google Scholar]
- 24.Greally MT. Shprintzen-Goldberg Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; Jan 13, 1993–2006. [updated Nov 16, 2010] [PubMed] [Google Scholar]
- 25.Maroteaux P, Le Merrer M. Maladies osseuses de l’ enfant. 4th ed. Paris: Medeicine-Sciences Flammarion; 2002. [Google Scholar]
- 26.De Backer J, Loeys B, Leroy B, Coucke P, Dietz H, De Paepe A. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin Genet. 2007;72(3):188–98. doi: 10.1111/j.1399-0004.2007.00845.x. [DOI] [PubMed] [Google Scholar]