

Abstract
AZ465 is a novel selective transient receptor potential cation channel, member A1 (TRPA1) antagonist identified during a focused drug discovery effort. In vitro, AZ465 fully inhibits activation by zinc, O-chlorobenzylidene malononitrile (CS), or cinnamaldehyde of the human TRPA1 channel heterologously expressed in human embryonic kidney cells. Our data using patch-clamp recordings and mouse/human TRPA1 chimeras suggest that AZ465 binds reversibly in the pore region of the human TRPA1 channel. Finally, in an ex vivo model measuring TRPA1 agonist-stimulated release of neuropeptides from human dental pulp biopsies, AZD465 was able to block 50%–60% of CS-induced calcitonin gene-related peptide release, confirming that AZ465 inhibits the native human TRPA1 channel in neuronal tissue.
Keywords: pain, pharmacology, antagonist, chimeric proteins, dental pulp, inflammation, neuropeptide, calcitonin gene-related peptide, CGRP
Introduction
The transient receptor potential cation channel, member A1 (TRPA1), is a cation-permeable channel belonging to the TRP channel superfamily, which has emerged as an important molecular target for several types of pain and inflammatory conditions. TRPA1 is highly expressed in sensory neurons in dorsal root ganglia and trigeminal ganglia, where it colocalizes with another TRP channel, TRPV1,1,2 and acts as a cellular sensor for various stimuli. TRPA1 is reported to be activated by noxious cold,3 some divalent ions (eg, Ca2+ Zn2+),4,5 and by a number of exogenous electrophilic compounds, such as the pungent ingredients in mustard oil (allyl isothiocyanate), ginger, cinnamon oil (cinnamaldehyde),6,7 garlic (allicin),8 and various tear gases9 (eg, O-chlorobenzylidene malononitrile [CS]), all of which can induce nocifensive behaviors, burning pain sensation, and sensory neuron sensitization in animals and man (see review in Radresa et al10). In addition to being gated by exogenous noxious chemicals, TRPA1 also responds to a variety of endogenous reactive compounds involved in oxidative stress and inflammation, such as 4-hydroxynonenal,11 hydrogen peroxide,12 and nitrooleic acid.13 Conversely, treatment with TRPA1 inhibitors reduces signs of hypersensitivity in various rodent models of inflammatory pain (see review in Radresa et al10). For instance, antagonism by the TRPA1 inhibitors HC-030031 and A-967079 completely blocked mechanical and cold hyperalgesia induced by intraplantar injection of Freund’s complete adjuvant14,15 and intrathecal injection of AP-18 reduced systemic hyperalgesia induced by the major proinflammatory cytokine tumor necrosis factor alpha (TNFα).16,17 Recently, the role of TRPA1 in pain was further strengthened by the discovery that a gain-of-function mutation of the channel is linked to a chronic human pain syndrome – familial episodic pain syndrome.18
The strength of evidence supporting TRPA1 as an attractive target against which to develop novel analgesics has led the pharmaceutical industry to invest in drug-discovery programs to identify TRPA1 antagonists. However, only a limited number of small molecules have been described in literature and patents so far. One possible contributing factor to the paucity of drug candidates is the cross-species difference in TRPA1 pharmacology; discrete variations in amino acid sequence results in key pharmacological and functional differences between TRPA1 orthologs. For instance, substances identified as antagonists at the human isoform showed no or opposite pharmacology at the rat receptor,19–21 highlighting the critical importance in drug-discovery programs aiming at TRPA1 of choosing the right surrogate species and developing ex vivo pharmacodynamic models using relevant human tissue.
In this respect, the dental pulp presents several advantages. Composed of various cell types, including odontoblasts, fibroblasts, inflammatory cells, vascular cells, and sensory neurons (unmyelinated C- and small-diameter myelinated AΔ-fibers containing neuropeptides such as substance P and calcitonin gene-related peptide [CGRP]), it constitutes a readily accessible neurological human tissue for ex vivo use that expresses TRPA1.22 Recently, Fehrenbacher et al23 developed an ex vivo model where CGRP release from human dental pulp following exposure to capsaicin was measured and could be blocked in a concentration-dependent manner by specific TRPV1 antagonists. Since TRPA1 activation has also been shown to trigger the release of neuropeptides from peripheral (meningeal) sensory neurons,24 we undertook to set up an adaptation of Fehrenbacher et al’s human dental pulp model23 for the study of TRPA1 antagonists.
AZ465, a novel potent TRPA1 antagonist, was identified during a focused drug-discovery program at AstraZeneca R&D (Södertälje, Sweden). We describe herein AZ465’s in vitro pharmacological profile and binding characteristics. In addition, our data using AZD465 suggest that measuring the release of CGRP from human dental pulp can be used as an ex vivo model system to study the inhibitory effects of TRPA1 antagonists in humans.
Materials and methods
Chemicals
AZ465 ((R)- N-(1-(4-cyanophenyl)-3,3-dimethyl-1-oxobutan-2-yl)-4-(6-methylpyridin-2-yl)piperazine-1-carboxamide; Figure 1) was synthesized at AstraZeneca R&D Södertälje (for details, see published patent application WO2012050512, where AZ465 is identified as “Formula [I]”).25
Figure 1.

Chemical structure of AZ465.
CS was manufactured at AstraZeneca Pharmaceutical Development (Bangalore, India). HC-030031 was purchased from TOSLab (Ekaterinburg, Russia). The Frescati antibody was manufactured by Agrisera (Vännäs, Sweden) for AstraZeneca exclusively.
Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMax, Hanks’ balanced salt solution with and without out Ca2+ and Mg2, and NaHCO3 were purchased from Life Technologies (Carlsbad, CA). Fetal calf serum (FCS) was purchased from HyClone (Logan, UT). Doxycycline was purchased from BD Biosciences (Franklin Lakes, NJ). Zinc chloride, HEPES (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid), trans-cinnamaldehyde, allyl isothiocyanate, NaOH, MgCl2, KCl, NaCl, and EGTA were purchased from Sigma-Aldrich (St Louis, MO). CaCl2 was purchased from Fluka (St Louis, MO). Fluo-4 NW was purchased from Life Technologies. Poly-d-lysine-coated 384-well plates were purchased from Falcon (Franklin Lakes, NJ). Polystyrene flat-bottom plates were purchased from Greiner Bio-One (Frickenhausen, Germany). Cell-culture dishes (60 mm) were purchased from Corning (Corning, NY).
Molecular cloning and cell-line generation
Full length c-DNA sequences based on Genebank NM_007332 (human TRPA1) were cloned into the pT-REx-DEST vector for tetracycline-inducible expression in a human embryonic kidney 293 tetracycline inducible cell line (HEK293-T-REx) (Life Technologies). For continuous culturing, TRPA1-HEK293-T-REx cells were grown as monolayers in medium (DMEM with GlutaMax, 10% FCS) supplemented with 400 μg/mL G418 and 1 μg/mL blasticidin.
Chimeras of mouse and human TRPA1 were designed and synthesized by GeneArt® (Life Technologies). The most common full-length cDNA sequences for mouse (NM_177781) and human (NM_007332) were chosen. The wild-type (WT) mouse and the chimeric mouse cDNA constructs with human exons 19–23 (TM1-6) or exons 22 and 23 (TM5 and -6) were murine codon-optimized and subcloned into pcDNA4TO vector (Life Technologies) from a pMK GeneArt vector. More specifically, the human exons 19–23 insert sequence stars at amino acid 713 and end at amino acid 959, and the human exons 22 and 23 start at amino acid 855, ending at the same position. Numbers correspond to the full-length mouse sequence NM_177781.
All constructs were verified by DNA sequencing. HEK-293T cells were transiently transfected (Lipofectamine 2000; Life Technologies) with the chimeric constructs 24–48 hours before the Ca2+ flux experiments.
Calcium-influx experiments
The calcium-influx measurements were done on a fluorescent imaging plate reader (FLIPR Tetra; Molecular Devices, Sunnyvale, CA). Stably expressing human TRPA1-HEK293-T-REx cells or transiently transfected chimeric mouse/human TRPA1-293T cells were resuspended in culture media (DMEM, 10% FCS). TRPA1 protein expressions in the TRPA1-HEK293-T-REx cells were induced by adding 1 μg/mL doxycyclin to the medium. Cells were seeded in 384-well poly-d-lysine-coated plates and incubated for 24–48 hours at 37°C, 5% CO2. Prior to the experiment, the medium was discarded and cells loaded with 25 μL per well Fluo-4 NW in assay buffer (Hanks’ balanced salt solution with Ca2+ and Mg2+, 0.4% NaHCO3, 10 mM HEPES, pH 7.4) for 1 hour at room temperature. AZ465 and the various agonists (zinc chloride, CS, and cinnamaldehyde) were serial diluted in 100% dimethyl sulfoxide (DMSO) and further diluted in assay buffer. The final DMSO concentration did not exceed 0.3% in any experiment. Validation experiments showed that concentrations below 0.9% DMSO did not interfere with the assay. For the agonist concentration-response experiments, an initial baseline measurement was made for 10 seconds followed by addition of 10–15 concentrations of zinc chloride, CS, or cinnamaldehyde. The antagonist-concentration response was assessed in a two-addition protocol. After an initial 10-second baseline measurement period, the test compound (antagonist) was added to the cell plate and incubated online for 5 minutes before the addition of an EC70–80 concentration of agonist. Raw fluorescent counts were measured for 10 minutes after last addition. Excitation LED banks with wavelength 470–495 nm and emission filters with wavelength 515–575 nm were used.
Patch-clamp recordings
Human TRPA1-HEK293-TREx cells were seeded on poly-d-lysine-coated coverslips in culture media (DMEM, 10% FCS) and induced with 1 μg/mL doxycyclin at 37°C and 5% CO2. Cells were used for patch-clamp experiments 48 hours after induction.
Whole-cell currents were measured using an EPC10 amplifier (Heka, Lambrecht/Pfalz, Germany) at room temperature (23°C). Pipette electrodes were made from borosilicate glass capillary tubes (GC150-10; Harvard Apparatus, Holliston, MA). The electrodes had resistances of 3–5 MΩ when filled with internal solution. The cell membrane was clamped to a potential of −80 mV and a 1-minute period was allowed to activate the TRPA1 channels after application of 250 μM cinnamaldehyde in the bath solution after rupture of the membrane. TRPA1 current traces were recorded before application of AZ465, in the presence of 30 μM AZ465, and after washout of AZ465. Between 50% and 80% of the series resistance was electronically compensated to minimize voltage errors, and currents were sampled at 10 kHz and filtered at 0.2 kHz. The ramp protocol consisted of a 500 ms ramp from −80 to +80 mV from a holding potential of 0 mV and was applied with a frequency of 1 Hz. The protocol was controlled and data were analyzed with PatchMaster version 2.20 (Heka).
The extracellular solution contained (in mM) 147 NaCl, 4 KCl, 10 glucose, 10 HEPES, 4 MgCl2, and 5 EGTA, and the pH was adjusted to 7.4 with NaOH. The intracellular solution contained (in mM) 120 CsCl, 10 EGTA, 10 HEPES, and 4 Na3GTP, and the pH was adjusted to 7.2 with CsOH.
Patients and ex vivo CGRP release from human dental pulp
The study was performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with good clinical practice and applicable regulatory requirements. Informed consent was obtained from patients prior to tooth extraction. Males and nonpregnant females from 18 years old were included in the study. All patients had healthy and normal pulp. Patients with bad oral status, HIV/hepatitis B/C, or other medical reasons for not being eligible, as judged by the investigator, were not included in the study.
Patients were anesthetized using nerve-block local injection with 3–4 mL 2% lidocaine/epinephrine. An ordinary surgical procedure was performed wherein a gingival flap was raised and bone was removed with a drill to get entrance for extraction of the tooth. The molar teeth where extracted or if necessary the crown was bisected from the roots with a drill before extraction. The coronal pulp was rapidly dissected and transferred to wells of a polypropylene 96-well plate containing ice-cold transplantation buffer (Aqix, London, UK). Each pulp was in turn divided in two or more parts (~5–15 mg each).
The ex vivo method, based on release of CGRP from dental pulp biopsies, is described elsewhere.23 However, minor adaptations are described below. Basal- and agonist-evoked peripheral release of CGRP were analyzed in the absence and presence of experimental pharmacological compounds according to the following sequence: (1) after overnight incubation on ice, tissue was washed once with medium and incubated for 30 minutes at 37°C; (2) the medium was changed, and samples for measurement of basal release were taken after 20 minutes; (3) 20-minute incubation with AZ465 or vehicle (solvent only) was followed by addition of agonist (final concentration, 200 μM CS) for another 20 minutes and samples corresponding to control and inhibited levels were obtained; and finally (4) the pulp tissue was incubated with capsaicin (50 μM, optimal concentration for maximal release23) for normalization of the release and to verify the responsiveness of the tissue. The final concentration of DMSO in the dental pulp assay did not exceed 0.5%. Tests made with dorsal root ganglion tissue showed that concentrations up to 1% DMSO did not result in altered CGRP release. Each sample was mixed with a 5× concentrate of EIA buffer (included in the kit) and frozen. The concentration of CGRP was measured by a commercial available immunoassay (SPI-Bio, Montigny le Bretonneux, France). In total, three samples were obtained from each piece of pulp tissue, and each pulp was used for analysis of control and at least one concentration of AZ465.
Tissue processing and staining
The presence of nociceptive markers of the dental pulp was analyzed by immunohistochemistry. Tissue was fixed in 4% paraformaldehyde (HL96753.1000; Histolab, Gothenburg, Sweden) for 72 hours, dehydrated for 13 hours, and finally embedded in paraffin. The dental pulp paraffin blocks were sectioned into 4 μm-thick sections using a microtome (HM 355s; Microm Internationatal, Walldorf, Germany).
TRPA1 immunohistochemistry was performed on Ventana Discovery XT using OmniMap DAB (760-159; Ventana, Tucson, AZ) detection. The sections were deparaffinized in EZ prep (950-100; Ventana), and antigen retrieval was achieved by incubation with cell-conditioning solution 1 (950-124; Ventana), a Tris ethylenediaminetetraacetic acid-based buffer (pH 7.8), for 48 minutes at 95°C. The TRPA1 (Frescati) antibody was diluted 1:100 (4 μg/mL) in antibody diluent (251-018; Ventana) and incubated for 60 minutes at room temperature. OmniMap anti-rabbit HRP-conjugated secondary antibody (760-4311; Ventana) was applied for 16 minutes at room temperature. The Frescati antibody was validated with Western blot and immunocytochemistry experiments on nontransfected and TRPA1-transfected HEK cells, as well as immunohistochemistry experiments on TRPA1-positive (tissue expressing TRPA1) and -negative (tissue not expressing TRPA1) tissue. Staining controls excluding primary and secondary antibody were also performed to exclude nonspecific staining (data not shown). The visualization was with 3,3′-diaminobenzidine (760-159; Ventana) incubated for 8 minutes at room temperature and counterstained with hematoxylin (760-2021; Ventana) for 4 minutes. Slides were dehydrated in ethanol and xylene and coverslipped in Eukitt (03989; Fluka).
Data analysis
In the calcium-influx experiments, inhibition and stimulation curves were analyzed by nonlinear regression analysis with variable slope on GraphPad Prism version 5.00 for Windows (GraphPad Software, La Jolla, CA) using the equation Y = bottom + (top – bottom)/(1 + 10(LogIC50 – X) × HillSlope).
The mean and standard error of three to six repeats at each concentration were plotted to obtain the half maximal effective concentration/half maximal inhibitory concentration (EC50/IC50) value. For agonist experiments, the data were normalized to the smallest and highest value for each data set. For antagonist experiments, the data were normalized to the response from the applied agonist (100 μM zinc or 10 nM CS or 50 μM cinnamaldehyde) in the absence of the antagonist.
In the tooth-pulp model, CS-induced CGRP release, in the presence or absence of the TRPA1 antagonist AZ465, was expressed as a percentage of the capsaicin-evoked level minus the basal release, and compared to the level in control tissue from the same pulp. The data were analyzed using paired Student’s t-test.
Results
AZ465 potently and selectively inhibits the human TRPA1 in vitro
A FLIPR calcium-influx assay was used to determine the ability of AZ465 to inhibit human TRPA1 in vitro. Both Zn2+ and CS have been shown to be potent activators of human TRPA1.5,9 In our hands, Zn2+ and CS evoked a concentration-dependent Ca2+ influx with EC50 values of 3 μM and 0.7 nM, respectively (data not shown). AZ465 appeared to be a very potent inhibitor of both Zn2+ (100 μM)- and CS (10 nM)-induced TRPA1 activation, with IC50 values of 85 nM and 20 nM, respectively (Figure 2).
Figure 2.

AZ465 is a potent inhibitor of Zn2+ and CS-induced TRPA1 activation measured in a calcium-influx assay on FLIPR.
Notes: AZ465 inhibition of Zn2+- and CS-induced intracellular Ca2+ increase (FLIPR) in HEK cells expressing the human TRPA1. AZ465 was added 5 minutes before addition of 100 μM zinc chloride or 10 nM CS. AZ465 fully inhibits the Zn2+- and CS-induced activation with IC50 values of 85 nM and 20 nM, respectively. Data are normalized to the maximum response of the agonist in the absence of AZ465 and represent the means ± standard error of four or five measurements.
Abbreviations: CS, O-chlorobenzylidene malononitrile; FLIPR, fluorescent imaging plate reader; IC50, half maximal inhibitory concentration; TRPA1, transient receptor potential cation channel, member A1.
AZ465 displayed a high degree of selectivity for TRPA1. No effect of AZ465 up to 100 μM could be observed on the responses of human TRPV1- and TRPM8-expressing cells to capsaicin or menthol, respectively (data not shown) (Table S1). Tested on a panel of >100 receptors, ion channels, nuclear hormone receptors, transporters, and enzymes (MDS Pharma Services) (Table S2), AZ465, at a concentration of 10 μM, did not interact significantly (≤50% inhibition or stimulation of binding), with most of the targets yielding a selectivity >100-fold for hTRPA1. Interaction was only observed with the dopamine transporter (DAT) (Ki = 3.02 μM) and vasopressin 1A (V1A) receptor (Ki = 1.69 μM), two targets with unlikely involvement in our test system (dental pulp).
AZ465 binds reversibly in the transmembrane region of human TRPA1
To further characterize AZ465, we generated two mTRPA1-hTRPA1 chimeras where either the whole transmembrane domain (T1–T6) or the pore region (T5, T6) in the mouse TRPA1 channel was humanized (Figure 3). The generated chimeras were expressed in HEK-293T cells and assayed in a calcium-influx FLIPR assay. Transfection efficiency in transient expression systems is usually lower than in stably expressing cells, and consequently the fluorescent response for the stable expressing cell line hTRPA1-WT was higher than for the three transiently transfected cell lines. Nevertheless, mTRPA1-WT, hTRPA-WT, and the two chimeras mTRPA1-hT1T6 and mTRPA1-hT5T6 were activated by cinnamaldehyde with similar EC50 values (Figure 4A), a response that could be blocked by the TRPA1 antagonist HC-030031, indicating that all constructs were functional (Figure 4C). When tested in these cell lines, AZ465 was able to block the cinnamaldehyde-induced response in the hTRPA1-WT (IC50 305 nM) but not in the mTRPA1-WT cells, in line with our observation that most of the compounds in AZ465’s chemical series lost their activity going from human to rodent TRPA1. Interestingly, when the transmembrane domain (T1–T6) in the mouse channel was humanized (mTRPA1-hT1T6), AZ465 ability to block the cinnamaldehyde response was recovered (IC50 370 nM). Replacing only the pore region (T5, T6) of the mouse channel with the human one (mTRPA1-hT5T6) resulted in a similar recovery (IC50 570 nM) (Figure 4B), indicating that amino acid residues within the pore region of the hTRPA1 channel are critical for the binding of AZ465. This finding is in line with the result of a recent study where a homology model of TRPA1 based on Kv1.2 was used to identify residues available for interaction with ligands entering the pore vestibule. Site-directed mutation constructs were expressed in Xenopus oocytes and their functionality and pharmacology assessed to validate the homology model. Based on the results, an antagonist-binding site in the pore vestibule of the TRPA1 ion channel was suggested for compounds within AZ465’s chemical series (Klement et al, unpublished data, 2012).
Figure 3.
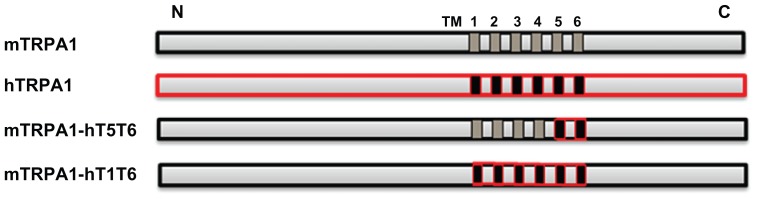
Schematic drawing of the TRPA1 constructs.
Notes: The transmembrane regions are indicated as TM1-6. The human sequences are indicated in red, both in the full-length and chimeric constructs. TM1-6 corresponds to exons 19–23 and TM5 and -6 to exons 22 and 23. For detailed information, see materials and methods.
Abbreviations: CS, O-chlorobenzylidene malononitrile; TRPA1, transient receptor potential cation channel, member A1.
Figure 4.

AZ465 inhibits cinnamaldehyde-induced TRPA1 activation in hTRPA1 and mouse/human chimeric TRPA1 channels, but not in mTRPA1. (A) Agonist concentration-response curves for cinnamaldehyde on hTRPA1-WT, mTRPA1-hT1T6, mTRPA1-hT5T6, and mTRPA1-WT in a Ca2+− influx assay. Cinnamaldehyde activates hTRPA1, mTRPA1-hT1T6, mTRPA1-hT5T6, and mTRPA1 with similar half maximal effective concentration (EC50)values (6.8 μM, 7.7 μM, 8.5 μM, and 6.9 μM, respectively). Data are plotted as peak fluorescent counts for each concentration and represent means ± standard of four or five measurements. Antagonist concentration-response curves for AZ465 (B) and HC-030031 (C) against cinnamaldehyde (33 μM)-induced TRPA1 activation.
Note: AZ465 fully inhibits the activation in hTRPA1, mTRPA1-hT1T6, and mTRPA1-hT5T6 with half maximal inhibitory concentration (IC50) values of 305 nM, 370 nM, and 570 nM, respectively, but AZ465 is inactive in the mTRPA1-WT cells. HC-030031 fully inhibits the activation in all cell lines, including mTRPA1-WT, with similar IC50 values (hTRPA1-WT, 10.5 μM; mTRPA1-hT1T6, 5.5 μM; mTRPA1-hT5T6, 9.5 μM; mTRPA1-WT 6.5 μM). Data are normalized to the maximum response evoked by 33 μM cinnamaldehyde in the absence of AZ465 or HC-030031 and represent the means ± standard of four or five measurements.
Abbreviations: TRPA1, transient receptor potential cation channel, member A1; WT, wild type.
To investigate the reversibility of AZ465 binding on TRPA1, whole-cell patch-clamp experiments were performed. Application of 30 μM AZ465 resulted in a rapid and complete inhibition of the cinnamaldehyde-activated TRPA1 currents, which recovered within seconds following washout (Figure 5).
Figure 5.

AZ465 reversibly inhibits cinnamaldehyde-activated TRPA1 currents in whole-cell patch-clamp experiments. (A) Cinnamaldehyde-activated TRPA1 current traces in response to a voltage ramp (−80 mV to +80 mV in 500 ms, Vhold = 0 mV) before application of AZ465 (control), in the presence of 30 μM AZ465 (AZ465), and after washout of AZ465 (recovery). (B) Time course of steady-state cinnamaldehyde-activated TRPA1 current amplitudes measured at +80 mV showing the reversible effect of repeated application and washout of AZ465. (C) AZ465 reduces the cinnamaldehyde-activated TRPA1 current to 5.4% ± 1.7% of the control current (n = 3, white bar).
Note: Washout of AZ465 leads to a full recovery of the TRPA1 response, with current amplitudes reaching 99.6% ± 8.9% of the control current (n = 3, black bar).
Abbreviations: TRPA1, transient receptor potential cation channel, member A1; WT, wild type.
Together, our findings suggest that (1) AZ465 binds to the transmembrane domain of the human TRPA1 channel, most likely the part that contains pore region; and (2) the mechanism of TRPA1 inhibition by AZ465 does not involve the formation of strong covalent bonds with the ion-channel protein.
AZ465 blocks CS-evoked CGRP release in human dental pulp ex vivo
In this study, we explored the ability of AZ465 to block CGRP release evoked by the potent TRPA1 agonist CS in human tooth pulp ex vivo. CS, a compound used as a tear gas for riot control, has been shown to be >100-fold more potent than cinnamaldehyde at gating TRPA1 in vitro, and they both do so by binding covalently in the cysteine-rich N-terminus. Because of its higher potency, CS was considered more suitable than cinnamaldehyde for ex vivo tissue experiments.
In total, 68 tooth-pulp samples were analyzed, corresponding to 33 teeth from 31 patients. In the first part of the study, the optimal concentration of CS was investigated. CS (1, 10, 25, 50, 100, 200, and 500 μM) caused a concentration-dependent increase in CGRP release that peaked at 200 μM. As a quality control of tissue responsiveness and viability and to normalize the data, 50 μM of capsaicin was added to the preparation at the end of each experiment. All samples with capsaicin-induced responses below 50 pg CGRP/mL were discarded, leaving 41 samples for further analysis.
AZ465 (10 and 50 μM) produced a significant inhibition of CS-evoked CGRP release in human dental pulp (Figure 6). The higher concentration (50 μM) of AZ465 was tested in six different individuals and resulted in an average ~2.5-fold reduction of CGRP release (P = 0.003, paired t-test). Application of 10 μM of AZ465 resulted in a ~2.0-fold reduction of CGRP release (P = 0.011, n = 4, paired t-test).
Figure 6.

Significant inhibition of CS-evoked CGRP release in human dental pulp by AZ465. Each pair of samples represents data from one pulp. Data from ten individuals are shown. Release of CGRP was expressed as percentage of maximum release obtained by capsaicin minus basal release from the tissue. The concentration of CS was 200 μM in all experiments; the concentrations of AZ465 were 10 μM (A) and 50 μM (B).
Abbreviations: CGRP, calcitonin gene-related peptide; CS, O-chlorobenzylidene malononitrile.
Also, the expression of TRPA1 in dental pulp samples was verified using immunohistochemistry (Figure 7). Our data confirm the expression of TRPA1 in human dental pulp, with immunopositive cells more frequently expressed in the peripheral parts of the tissue, in line with a recent report.22
Figure 7.

TRPA1 immunohistochemistry in dental pulp.
Note: Representative image showing the expression patterns of TRPA1 (brown) in the human coronal dental pulp.
Abbreviation: TRPA1, transient receptor potential cation channel, member A1.
Discussion
In this paper, we have provided evidence that the novel compound AZ465 is a potent and selective TRPA1 antagonist with activity in native human tissue.
In vitro, as measured in a Ca2+ flux assay, AZ465 fully inhibited the TRPA1 response to cinnamaldehyde and CS, two highly electrophilic compounds that activate hTRPA1 through covalent cysteine binding.9,26,28 AZ465 also fully inhibited TRPA1 activation by Zn2+, which most likely occurs via a different mechanism involving interaction with an intracellular C-terminal site.5,27 The IC50 values for AZ465 varied somewhat depending on the agonist (30 ± 10 nM vs CS, 85 ± 20 nM vs Zn2+, 305 ± 65 nM vs cinnamaldehyde, mean ± standard error, n = 4). One possible contributing factor was that the concentrations of the agonists used in the assay were an estimation of their EC70–80 and even though our data show that AZ465 binds in the transmembrane region of the channel, suggesting a noncompetitive interaction between AZ465 and the agonists, some shift in the inhibition dose-response curve for AZ465 depending on the agonist concentration could nonetheless be seen for cinnamaldehyde and CS (data not shown). Still, our results indicate that AZ465 is effective in blocking TRPA1 regardless of the gating modality. AZ465 also appeared to be highly selective for human TRPA1, with no affinity for hTRPM8, hTRPV1, or 104 of the 106 other molecular targets tested, with only moderate affinity for two – DAT and V1A – to our knowledge not expressed in peripheral sensory neurons.
Using patch-clamp experiments and mouse/human TRPA1 chimeras, we were able to demonstrate that AZ465 binds reversibly to the pore region (T5, T6) of the human but not the mouse (or the rat, data not shown) channel. This finding is not surprising considering the limited sequence identity between human and rodent TRPA1 proteins (about 80%). Also, amino acids in the T5/T6 region are known to play a key role in mediating the response of TRPA1 to pharmacological tools. For instance, the electrophilic antagonist CMP1 acted as an agonist when the pore region (T5, T6) of the human protein was exchanged for the rat sequence. Conversely, CMP1 acted as an antagonist at the rat TRPA1 when the pore region was “humanized,” specific residues in T6 being critical for the difference.29 Similarly, key residues in the T5 region of TRPA1 point towards this region as the critical domain for the antagonistic action of menthol.21
Given that discrete variations in amino acid sequence may be responsible for pharmacological and functional differences between TRPA1 orthologs, confirming the pharmacological activity of compounds in native human tissue is essential in a drug-discovery program. Therefore, we have developed an ex vivo pharmacodynamic model using human dental pulp to characterize our TRPA1 antagonists.
Previously, capsaicin had been shown to evoke CGRP release in a human dental pulp preparation, a response that could be fully inhibited by the TRPV1 antagonist capsazepine.23 Using an adaptation of the method, we show here that CS triggers the release of CGRP from human dental pulp biopsies, a response that could be reduced by AZD465, indicating its ability to inhibit the native human TRPA1 channel.
Even though CS is a very potent TRPA1 activator on recombinant TRPA1 cells in vitro, micromolar concentrations were needed to elicit a robust CGRP release in the pulp. It is indeed a common observation that when going from a cell culture to a complex tissue, higher concentrations of a ligand are required to evoke a pharmacological response. For instance, in the same preparation, micromolar concentrations of the TRPV1 agonist capsaicin were needed to trigger the release of neuropeptides (EC50 ~20 μM),23 although capsaicin is a far more potent TRPV1 activator in vitro in cell preparations with an EC50 in the low nanomolar range.30 In our dental pulp preparation, 10 mM of cinnamaldehyde (maximum concentration in an acceptable vehicle) triggered only low levels of CGRP release (data not shown), and CS was selected instead.
Another observation was that the CGRP release was only reduced by 50%–60% at the highest concentration of AZ465. Several explanations may account for this partial inhibition. CS is a reactive compound like most of the known TRPA1 agonists, activating the channel through chemical reactivity of their electrophilic groups with cysteine residues in the protein, and as such it can readily react with nucleophilic sites in different enzymes and other proteins.31 Therefore, it cannot be excluded that at the concentrations used here, CS reacts with other targets, of which some could trigger a downstream CGRP release independent of TRPA1, and as such, is insensitive to AZ465. Also, as discussed above for CS, the effective concentration of AZ465 that actually reached TRPA1 within the complex, nondissociated pulp tissue was most likely lower than the concentrations (10 and 50 μM) applied to the preparation. Like many drug-like small molecules, AZ465 binds nonspecifically to other proteins, like albumin, present in blood and tissues (AZ465 plasma protein binding ~98%, data not shown) reducing the free fraction available for interaction with TRPA1. Due to AZ465’s limited water solubility, testing higher concentrations than 50 μM to address whether CGRP release could be fully blocked was not feasible in an acceptable vehicle.
Finally, another possible hypothesis for the partial inhibition could be that the native target expressed in the human dental pulp is different from the recombinant TRPA1 expressed in the HEK cells, and that AZ465 doesn’t fully block native TRPA1 activation. Structurally, TRPA1 are tetramers and form homo- or heteromeric complexes. For instance, TRPA1 has been reported to colocalize with TRPV11,32 in dorsal root ganglia and trigeminal neurons. Functionally, it has been demonstrated that coexpression of TRPA1 with TRPV1 alters single-channel properties for AITC as well as the pharmacology for TRPA1 agonists and antagonists.2,32,33 It cannot be excluded that such cross-modulation of TRPA1 biophysical and regulatory properties or other post translational modifications could alter the pharmacology of AZ465 in native tissue as compared to the pharmacology in a recombinant system.
In summary, although additional studies using alternative agonists and antagonists, prolonged incubation times, etc, are necessary to complete its validation, the dental pulp model is a promising approach for testing the pharmacology of TRPA1 antagonists at the native human channel.
Conclusion
AZ465 is a novel, potent, selective, and fully reversible inhibitor of human TRPA1 in in vitro recombinant systems and is also active on the native human TRPA1. Our model of CS-induced CGRP release from human dental pulp represents a promising translational tool for the pharmacological characterization of TRPA1 antagonists in human native tissue.
Supplementary materials
Table S1.
Selectivity targets tested at AstraZeneca at concentration 100 μM
| Ligand-gated ion channels | TRPV1 | Rat |
| Ligand-gated ion channels | TRPV1 | Human |
| Ligand-gated ion channels | TRPM8 | Human |
| Enzymes | Prostaglandin E synthases 1 (PGES1) | Human |
| Voltage-gated ion channels | NaV1.7 | Human |
Table S2.
Selectivity targets tested at MDS Pharma Services at concentration 10 μM
| Target class | Target | Species |
|---|---|---|
| Enzymes | Monoamine oxidase, MAOA | Human |
| Enzymes | Nitric oxide synthase, endothelial eNOS | Bovine |
| Enzymes | Angiotensin converting enzyme | Rabbit |
| Enzymes | Acetylcholinesterase | Human |
| Enzymes | Aldose reductase | Rat |
| Enzymes | Cathepsin B | Human |
| Enzymes | Matrix metalloproteinase (MMP)-2 | Human |
| Enzymes | Cyclooxygenase, COX-2 | Human |
| Enzymes | Cyclooxygenase, COX-1 | Human |
| Enzymes | Renin | Human |
| Enzymes | Phosphodiesterase PDE 3 | Human |
| Enzymes | Thrombin | Human |
| Enzymes | ATPase, Na+/K+ | Porcine |
| Enzymes | MMP-14 | Human |
| Enzymes | Caspase 1 | Human |
| Enzymes | Peptidase, endothelin converting enzyme (ECE)-1 | Human |
| Enzymes | Protein tyrosine phosphatase, PTPRC | Human |
| Enzymes | Nitric oxide synthase, endothelial eNOS | Bovine |
| Enzymes | Carbonic anhydrase II | Human |
| GPCR | Opiate, delta (OP1, DOP) | Human |
| GPCR | Opiate, mu (OP3, MOP) | Human |
| GPCR | Opiate, kappa (OP2, KOP) | Human |
| GPCR | Adrenergic, beta2 | Human |
| GPCR | Cannabinoid, CB2 | Human |
| GPCR | Adenosine, A2A | Human |
| GPCR | Adrenergic, alpha1A | Rat |
| GPCR | Adenosine, A1 | Human |
| GPCR | Adenosine, A3 | Human |
| GPCR | Adrenergic, alpha2A | Human |
| GPCR | Adrenergic, beta1 | Human |
| GPCR | Adrenergic, alpha2B | Human |
| GPCR | Endothelin, ETA | Human |
| GPCR | Dopamine, D4.2 | Human |
| GPCR | Dopamine, D2L | Human |
| GPCR | Dopamine, D1 | Human |
| GPCR | Cholecystokinin CCK2 (CCKB) | Human |
| GPCR | Serotonin, 5-HT4 | Guinea pig |
| GPCR | Prostanoid, thromboxane A2 | Human |
| GPCR | Histamine, H2 | Human |
| GPCR | Chemokine CXCR2 (IL-8B) | Human |
| GPCR | Cysteinyl CysLT1 | Human |
| GPCR | Melanocortin MC4 | Human |
| GPCR | Motilin | Human |
| GPCR | Muscarinic, M2 | Human |
| GPCR | Muscarinic, M3 | Human |
| GPCR | Neuropeptide, Y1 | Human |
| GPCR | Serotonin, 5-HT1A | Human |
| GPCR | Serotonin (5-hydroxytryptamine) 5-HT2A | Human |
| GPCR | Serotonin (5-hydroxytryptamine) 5-HT2B | Human |
| GPCR | Vanilloid | Rat |
| GPCR | Platelet activating factor (PAF) | Human |
| GPCR | Growth hormone secretagogue (GHS, Ghrelin) | Human |
| GPCR | Histamine, H1 | Human |
| GPCR | Bombesin BB2 | Human |
| GPCR | Adrenergic, alpha1D | Human |
| GPCR | Gamma-aminobutyric acid (GABA) B1a | Human |
| GPCR | Melatonin receptor 1A, MT1 | Human |
| GPCR | Serotonin, 5-HT1B | Rat |
| GPCR | Cholecystokinin CCK1 (CCKA) | Human |
| GPCR | Somatostatin sst4 | Human |
| GPCR | Cannabinoid, CB1 | Human |
| GPCR | Chemokine, CCR1 | Human |
| GPCR | Adenosine, A2A | Human |
| GPCR | Bradykinin, B2 | Human |
| GPCR | Calcitonin gene-related peptide CGRP1 | Human |
| GPCR | Tachykinin, NK1 | Human |
| GPCR | Neurotensin receptor 1 | Human |
| GPCR | Leukotriene B4 receptor | Human |
| GPCR | P2Y purine receptor | Rat |
| Kinases | Rho-associated, coiled-coil containing protein kinase 1 (ROCK1) | Human |
| Kinases | Fibroblast growth factor receptor 1 (FGFR1) | Human |
| Kinases | Vascular endothelial growth factor (VEGFR), cFit | Human |
| Kinases | Hepatocyte growth factor receptor (HGFR) (cMet) | Human |
| Kinases | Tyrosine-protein kinase Yes | Human |
| Kinases | Epidermal growth factor receptor (EGFR) | Human |
| Kinases | Insulin receptor | Human |
| Kinases | Sarcoma (Src) | Human |
| Kinases | Fyn-related kinase (Fyn) | Human |
| Ligand-gated ion channels | Nicotinic acetylcholine | Human |
| Ligand-gated ion channels | Glycine, strychnine-sensitive | Rat |
| Ligand-gated ion channels | Glutamate, NMDA, agonism | Rat |
| Ligand-gated ion channels | Glutamate, NMDA, phencyclidine | Rat |
| Ligand-gated ion channels | Glutamate, NMDA, glycine | Rat |
| Ligand-gated ion channels | Serotonin, 5-HT3 | Human |
| Ligand-gated ion channels | GABAA, benzodiazepine, central | Rat |
| Nuclear receptors | Estrogen, ERa | Human |
| Nuclear receptors | Testosterone | Rat |
| Nuclear receptors | Progesterone PRB | Human |
| Nuclear receptors | Glucocorticoid | Human |
| Nuclear receptors | Retinoid X receptor RXRa | Human |
| Other receptors | Sigma 1 | Human |
| Other receptors | Benzodiazepine, peripheral | Rat |
| Receptor tyrosine kinases | Epidermal growth factor | Human |
| Transporters | Adenosine transporter | Guinea pig |
| Transporters | Noradrenaline transporter | Human |
| Transporters | Dopamine transporter | Human |
| Transporters | Serotonin transporter | Human |
| Voltage-gated ion channels | KATP channel | Hamster |
| Voltage-gated ion channels | Calcium channel, L-type, dihydropyridine | Rat |
| Voltage-gated ion channels | Calcium channel, L-type, benzothiazepine | Rat |
| Voltage-gated ion channels | Human ether-à-go-go-related gene (hERG) | Human |
| Voltage-gated ion channels | NaV1.5 | Human |
| Voltage-gated ion channels | Ito Kv4.3 potasssium channel | Human |
| Voltage-gated ion channels | IKs potassium channel | Human |
| Voltage-gated ion channels | CaN N-type calcium channel | Rat |
Acknowledgment
The authors thank Dr Steven England for sharing his vast knowledge of ion channel science and for his guidance.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Kobayashi K, Fukuoka T, Obata K, et al. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J Comp Neurol. 2005;493:596–606. doi: 10.1002/cne.20794. [DOI] [PubMed] [Google Scholar]
- 2.Salas M, Hargreaves K, Akopian A. TRPA1-mediated responses in trigeminal sensory neurons: interaction between TRPA1 and TRPV1. Eur J Neurosci. 2009;29:1568–1578. doi: 10.1111/j.1460-9568.2009.06702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Story G, Peier A, Reeve A, et al. ANKTM1, a TRP-like Channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 4.Doerner JF, Gisselmann G, Hatt H, Wetzel CH. Transient receptor potential channel A1 is directly gated by calcium ions. Biol Chem. 2007;282:13180–13189. doi: 10.1074/jbc.M607849200. [DOI] [PubMed] [Google Scholar]
- 5.Hu H, Bandell M, Petrus MJ, Zhu MX, Patapoutian A. Zinc activates damage-sensing TRPA1 ion channels. Nat Chem Biol. 2009;5:183–190. doi: 10.1038/nchembio.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jordt SE, Bautista DM, Chuang HH, et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 7.Bandell M, Story GM, Hwang SW, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 8.Macpherson LJ, Geierstanger BH, Viswanath V, et al. The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr Biol. 2005;15:929–934. doi: 10.1016/j.cub.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Brône B, Peeters PJ, Marrannes R, et al. Tear gasses CN, CR, and CS are potent activators of the human TRPA1 receptor. Toxicol Appl Pharmacol. 2008;231:150–156. doi: 10.1016/j.taap.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Radresa O, Dahllöf H, Nyman E, et al. TRPA1 in pain pathophysiology and implications for the development of a new class of analgesic drugs. Open Pain J. 2012 In press. [Google Scholar]
- 11.Trevisani M, Siemens J, Materazzi S, et al. 4-hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A. 2007;104:13519–13524. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawada Y, Hosokawa H, Matsumura K, Kobayashi S. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci. 2008;27:1131–1142. doi: 10.1111/j.1460-9568.2008.06093.x. [DOI] [PubMed] [Google Scholar]
- 13.Taylor-Clarke TE, Ghatta S, Bettner W, Undem BJ. Nitrooleic ocid, an endogenous product of nitrative stress, activates nociceptive sensory neurons via the direct activation of TRPA1. Mol Pharmacol. 2009;75:820–829. doi: 10.1124/mol.108.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrus M, Peier AM, Bandell M, et al. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain. 2007;3:40. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eid SR, Crown ED, Moore EL, et al. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain. 2008;4:48. doi: 10.1186/1744-8069-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandes ES, Russell FA, Spina D, et al. A distinct role for transient receptor potential ankyrin 1, in addition to transient receptor potential vanilloid 1, in tumor necrosis factor alpha-induced inflammatory hyperalgesia and Freund’s complete adjuvant-induced monarthritis. Arthritis Rheum. 2011;63:819–829. doi: 10.1002/art.30150. [DOI] [PubMed] [Google Scholar]
- 17.McGaraughty S, Chu KL, Perner RJ, Didomenico S, Kort ME, Kym PR. TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Mol Pain. 2010;6:14. doi: 10.1186/1744-8069-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kremeyer B, Lopera F, Cox JJ, et al. A gain-of-function mutation in TRPA1 causes familiar episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky L, Tamir R, Gao B, et al. Species-specific pharmacology of Trichloro(sulfanyl)ethyl benzamides as transient receptor potential ankyrin 1 (TRPA1) antagonists. Mol Pain. 2007;3:39. doi: 10.1186/1744-8069-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagatomo K, Ishii H, Yamamoto T, Nakajo K, Kubo Y. The Met268Pro mutation of mouse TRPA1 changes the effect of caffeine from activation to suppression. Biophys J. 2010;99:3609–3618. doi: 10.1016/j.bpj.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao B, Dubin AE, Bursulaya B, Viswanath V, Jegla TJ, Patapoutian A. Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J Neurosci. 2008;28:9640–9651. doi: 10.1523/JNEUROSCI.2772-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YS, Jung HK, Kwon TK, et al. Expression of transient receptor potential ankyrin in human dental pulp. J Endod. 2012;38:1087–1092. doi: 10.1016/j.joen.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 23.Fehrenbacher JC, Sun XX, Locke EE, Henry MA, Hargreaves KM. Capsaicin-evoked iCGRP release from human dental pulp: a model system for the study of peripheral neuropeptide secretion in normal healthy tissue. Pain. 2009;144:253–261. doi: 10.1016/j.pain.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunkler PE, Ballard CJ, Oxford GS, Hurley JH. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain. 2011;152:38–44. doi: 10.1016/j.pain.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weigelt D, Svensson M. Patent application WO 2012050512. TRPA1 receptor antagonist. 2012 Apr 19;
- 26.Bessac BF, Sivula M, von Hehn CA, Caceres AI, Escalera J, Jordt SE. Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. FASEB J. 2009;23:1102–1114. doi: 10.1096/fj.08-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banke TG, Wickenden AD. Intracellular zinc irritates TRPA1. Nat Chem Biol. 2009;5:141–142. doi: 10.1038/nchembio0309-141. [DOI] [PubMed] [Google Scholar]
- 28.Macpherson LJ, Dubin AE, Evans MJ, et al. Noxious compounds activate TRPA1 ion channels through covalent modifications of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Zhang XF, Kort ME, et al. Molecular determinants of species-species activation or blockade of TRPA1 channels. J Neurosci. 2008;28:5063–5071. doi: 10.1523/JNEUROSCI.0047-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC). 5th ed. Br J Pharmacol. 2011;164(Suppl 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blain PG. Tear gases and irritant incapacitants 1-chloroacetophenone, 2-chlorobenzylidene malononitrileand dibenz[B,F]-1,4-oxazepine. Toxicol Rev. 2003;22:103–110. doi: 10.2165/00139709-200322020-00005. [DOI] [PubMed] [Google Scholar]
- 32.Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPA1-TRPV1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem. 2010;285:15167–15177. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akopian AN. Nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr Pharm Biotechnol. 2011;12:89–94. doi: 10.2174/138920111793937952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Selectivity targets tested at AstraZeneca at concentration 100 μM
| Ligand-gated ion channels | TRPV1 | Rat |
| Ligand-gated ion channels | TRPV1 | Human |
| Ligand-gated ion channels | TRPM8 | Human |
| Enzymes | Prostaglandin E synthases 1 (PGES1) | Human |
| Voltage-gated ion channels | NaV1.7 | Human |
Table S2.
Selectivity targets tested at MDS Pharma Services at concentration 10 μM
| Target class | Target | Species |
|---|---|---|
| Enzymes | Monoamine oxidase, MAOA | Human |
| Enzymes | Nitric oxide synthase, endothelial eNOS | Bovine |
| Enzymes | Angiotensin converting enzyme | Rabbit |
| Enzymes | Acetylcholinesterase | Human |
| Enzymes | Aldose reductase | Rat |
| Enzymes | Cathepsin B | Human |
| Enzymes | Matrix metalloproteinase (MMP)-2 | Human |
| Enzymes | Cyclooxygenase, COX-2 | Human |
| Enzymes | Cyclooxygenase, COX-1 | Human |
| Enzymes | Renin | Human |
| Enzymes | Phosphodiesterase PDE 3 | Human |
| Enzymes | Thrombin | Human |
| Enzymes | ATPase, Na+/K+ | Porcine |
| Enzymes | MMP-14 | Human |
| Enzymes | Caspase 1 | Human |
| Enzymes | Peptidase, endothelin converting enzyme (ECE)-1 | Human |
| Enzymes | Protein tyrosine phosphatase, PTPRC | Human |
| Enzymes | Nitric oxide synthase, endothelial eNOS | Bovine |
| Enzymes | Carbonic anhydrase II | Human |
| GPCR | Opiate, delta (OP1, DOP) | Human |
| GPCR | Opiate, mu (OP3, MOP) | Human |
| GPCR | Opiate, kappa (OP2, KOP) | Human |
| GPCR | Adrenergic, beta2 | Human |
| GPCR | Cannabinoid, CB2 | Human |
| GPCR | Adenosine, A2A | Human |
| GPCR | Adrenergic, alpha1A | Rat |
| GPCR | Adenosine, A1 | Human |
| GPCR | Adenosine, A3 | Human |
| GPCR | Adrenergic, alpha2A | Human |
| GPCR | Adrenergic, beta1 | Human |
| GPCR | Adrenergic, alpha2B | Human |
| GPCR | Endothelin, ETA | Human |
| GPCR | Dopamine, D4.2 | Human |
| GPCR | Dopamine, D2L | Human |
| GPCR | Dopamine, D1 | Human |
| GPCR | Cholecystokinin CCK2 (CCKB) | Human |
| GPCR | Serotonin, 5-HT4 | Guinea pig |
| GPCR | Prostanoid, thromboxane A2 | Human |
| GPCR | Histamine, H2 | Human |
| GPCR | Chemokine CXCR2 (IL-8B) | Human |
| GPCR | Cysteinyl CysLT1 | Human |
| GPCR | Melanocortin MC4 | Human |
| GPCR | Motilin | Human |
| GPCR | Muscarinic, M2 | Human |
| GPCR | Muscarinic, M3 | Human |
| GPCR | Neuropeptide, Y1 | Human |
| GPCR | Serotonin, 5-HT1A | Human |
| GPCR | Serotonin (5-hydroxytryptamine) 5-HT2A | Human |
| GPCR | Serotonin (5-hydroxytryptamine) 5-HT2B | Human |
| GPCR | Vanilloid | Rat |
| GPCR | Platelet activating factor (PAF) | Human |
| GPCR | Growth hormone secretagogue (GHS, Ghrelin) | Human |
| GPCR | Histamine, H1 | Human |
| GPCR | Bombesin BB2 | Human |
| GPCR | Adrenergic, alpha1D | Human |
| GPCR | Gamma-aminobutyric acid (GABA) B1a | Human |
| GPCR | Melatonin receptor 1A, MT1 | Human |
| GPCR | Serotonin, 5-HT1B | Rat |
| GPCR | Cholecystokinin CCK1 (CCKA) | Human |
| GPCR | Somatostatin sst4 | Human |
| GPCR | Cannabinoid, CB1 | Human |
| GPCR | Chemokine, CCR1 | Human |
| GPCR | Adenosine, A2A | Human |
| GPCR | Bradykinin, B2 | Human |
| GPCR | Calcitonin gene-related peptide CGRP1 | Human |
| GPCR | Tachykinin, NK1 | Human |
| GPCR | Neurotensin receptor 1 | Human |
| GPCR | Leukotriene B4 receptor | Human |
| GPCR | P2Y purine receptor | Rat |
| Kinases | Rho-associated, coiled-coil containing protein kinase 1 (ROCK1) | Human |
| Kinases | Fibroblast growth factor receptor 1 (FGFR1) | Human |
| Kinases | Vascular endothelial growth factor (VEGFR), cFit | Human |
| Kinases | Hepatocyte growth factor receptor (HGFR) (cMet) | Human |
| Kinases | Tyrosine-protein kinase Yes | Human |
| Kinases | Epidermal growth factor receptor (EGFR) | Human |
| Kinases | Insulin receptor | Human |
| Kinases | Sarcoma (Src) | Human |
| Kinases | Fyn-related kinase (Fyn) | Human |
| Ligand-gated ion channels | Nicotinic acetylcholine | Human |
| Ligand-gated ion channels | Glycine, strychnine-sensitive | Rat |
| Ligand-gated ion channels | Glutamate, NMDA, agonism | Rat |
| Ligand-gated ion channels | Glutamate, NMDA, phencyclidine | Rat |
| Ligand-gated ion channels | Glutamate, NMDA, glycine | Rat |
| Ligand-gated ion channels | Serotonin, 5-HT3 | Human |
| Ligand-gated ion channels | GABAA, benzodiazepine, central | Rat |
| Nuclear receptors | Estrogen, ERa | Human |
| Nuclear receptors | Testosterone | Rat |
| Nuclear receptors | Progesterone PRB | Human |
| Nuclear receptors | Glucocorticoid | Human |
| Nuclear receptors | Retinoid X receptor RXRa | Human |
| Other receptors | Sigma 1 | Human |
| Other receptors | Benzodiazepine, peripheral | Rat |
| Receptor tyrosine kinases | Epidermal growth factor | Human |
| Transporters | Adenosine transporter | Guinea pig |
| Transporters | Noradrenaline transporter | Human |
| Transporters | Dopamine transporter | Human |
| Transporters | Serotonin transporter | Human |
| Voltage-gated ion channels | KATP channel | Hamster |
| Voltage-gated ion channels | Calcium channel, L-type, dihydropyridine | Rat |
| Voltage-gated ion channels | Calcium channel, L-type, benzothiazepine | Rat |
| Voltage-gated ion channels | Human ether-à-go-go-related gene (hERG) | Human |
| Voltage-gated ion channels | NaV1.5 | Human |
| Voltage-gated ion channels | Ito Kv4.3 potasssium channel | Human |
| Voltage-gated ion channels | IKs potassium channel | Human |
| Voltage-gated ion channels | CaN N-type calcium channel | Rat |