

Abstract
Molecular characterization of subsurface microbial communities in the former Homestake gold mine, South Dakota, was carried out by 16S rDNA sequence analysis using a water sample and a weathered soil–like sample. Geochemical analyses indicated that both samples were high in sulfur, rich in nitrogen and salt, but with significantly different metal concentrations. Microbial diversity comparisons unexpectedly revealed three distinct operational taxonomic units (OTUs) belonging to the archaeal phylum Thaumarchaeota typically identified from marine environments, and one OTU to a potentially novel phylum that falls sister to Thaumarchaeota. To our knowledge this is only the second report of Thaumarchaeota in a terrestrial environment. The majority of the clones from Archaea sequence libraries fell into two closely related OTUs and grouped most closely to an ammonia–oxidizing, carbon–fixing and halophilic thaumarchaeote genus, Nitrosopumilus. The two samples showed neither Euryarchaeota nor Crenarchaeota members that were often identified from other subsurface terrestrial ecosystems. Bacteria OTUs containing the highest percentage of sequences were related to sulfur-oxidizing bacteria of the orders Chromatiales and Thiotrichales. Community members of Bacteria from individual Homestake ecosystems were heterogeneous and distinctive to each community with unique phylotypes identified within each sample.
Keywords: 16S rDNA, Thaumarchaeota, phylogenetic microbial diversity, Homestake gold mine, extremophiles
Introduction
Subsurface environments offer unique habitats to microorganisms, shielding them from cosmic rays and fluctuating temperature on the Earth's surface. However, nutritional limitation and extreme geochemical and physical conditions in deep environments lead to ecological isolation of microbial niches and subsequent genetic drift. In particular, the size and composition of the subsurface microbial communities are influenced by surrounding geological conditions such as rock chemistry, thermogradients, metal concentrations, and extreme pH, similar to those of early Earth or even outside the Earth [1]. These physical and chemical attributes of subterranean ecosystems may have caused the evolution of unique lineages within individual microbial communities. Equally, microbial metabolic activities have also cast the physicochemical properties of the surrounding geosphere, supporting the view of coevolution of life and the Earth [2].
The Homestake gold mine, located in the Black Hills, Lead, SD (44°35′2074″N, 103°75′082″W) is one of the deepest mines in the western hemisphere. Gold was produced from as deep as 2.5 km below the surface during its 125–year life until production ceased in 2001. Gold deposits in the mine typically occur in an iron formation known as the Homestake Formation, which was deposited approximately 1.9 b.y.a. during the Precambrian Era and metamorphosed about 1.7 b.y.a., although some gold was also found associated with Tertiary–age intrusive rocks [3]. The northern Black Hills underwent extensive intrusion of shallow–level igneous rocks from ~58 m.y.a. to ~46 m.y.a. [4]. Currently, the Homestake mine is being converted to a Deep Underground Science and Engineering Laboratory (DUSEL) with support from the National Science Foundation and other federal, state, and private agencies. Abundant information is available for the geology and related mining history of the Homestake gold mine [5-8]. However, data on microbiology is extremely limited with investigation on geomicrobiology being pursued only recently [9,10] . Recognizing that microorganisms are ubiquitous and play a critical role as catalysts in biogeochemical cycling, it is expected that microorganisms occupy unique ecological niches within the Homestake mine. During active mining operations, diverse surface microorganisms were likely introduced, allowing for interaction among themselves or with indigenous members through horizontal gene transfer [11-13]. Such microbial interactions under extreme environmental conditions result in genetically distinct extremophiles with diverse and unique metabolic features. Novel metabolic products from DUSEL extremophiles may offer significant potential for industrial, pharmaceutical, or environmental applications.
Recently, both culture–dependent and culture–independent techniques have been used to explore the microbial communities of globally distributed terrestrial extreme subsurface environments [14-16]. Extensive studies on deep boreholes in the mines in South Africa [17,18], a deep igneous rock aquifer in Finland [19], and Henderson Mine, Colorado, USA [20] have described unique microbial communities established under the pristine subsurface conditions and their interactions with surroundings. While surface and near–surface microorganisms rely on photosynthesis–derived organic carbon, deep–subsurface microorganisms utilize photosynthesis–independent energy sources, typically H2–based energy [21,22]. The geologic formations of the Homestake mine consist of metamorphic rocks that not only lack organic matter but have also been exposed to high temperature and pressure during their evolution [3]. Shortage of organic energy sources available to the geosphere at the deeper levels of the Homestake DUSEL may favor the growth of chemolithoautotrophic extremophiles with novel metabolic activities. In the absence of human activities, microorganisms in the pristine subsurface areas would have sustained their physiological novelty under such stringent environmental conditions. However, as evidenced in deep mines in South Africa where microbial contamination resulted due to anthropogenic activities [23], some degree of microbial diversity influenced by surface microbiota is expected in the deeper areas (down to 2.4 km) at Homestake DUSEL where mining operations were once active.
In 2001, mining activities at Homestake mine ceased. By 2002 the access shafts were sealed, ventilation of the drifts ceased and the water level began to rise as pumping of the subsurface aquifers ended. Over the next five years the subsurface ecosystem was left untouched by human activities, and there was no new introduction of surface microbes to the subsurface drifts and access shafts. In this study, we explore the microbial diversity within two samples of opportunity, water and soil, taken from the Homestake mine during early mine re–entry activities shortly after opening the Ross Shaft, and prior to re-establishment of ventilation and human access. Geochemistry of the samples was characterized using standard analysis methods, and the microbiology was characterized using a culture–independent method to compare the microbial community structures of the two subsurface ecosystems. Herein we describe the composition of archaeal and bacterial communities in each ecosystem based on 16S rDNA sequences in association with the geochemical characteristics of their surroundings.
Materials and methods
Sites and sampling
Main sampling activities for this study were coordinated with the re–entry schedule of the Homestake mine for its inspection by the South Dakota Science and Technology Authority (SDSTA) in 2007 prior to its conversion into a DUSEL. Upon opening of the Ross Shaft in May 2007, the water sample was collected in 5 sterile 1L polypropylene bottles along the length of a single seep running down from the 244–m to 914–m level of the Ross shaft. In September 2007, weathered soil–like material, herein referred to as soil, was taken from the surface of a stope at the horizontal drift, approximately 850 m off the Ross shaft at 610 m below the surface (2,000 ft. level). Figure 1a depicts a cross section view of the Homestake mine and the range of water sampling along the Ross shaft. Figure 1b is a detailed map including distribution of rock formations of the soil sampling site and adjacent areas in a drift at the 610 m level of the mine. All samples were taken in one–liter acid–washed, sterile polypropylene bottles and kept on ice while transported to the laboratory and stored at 4°C while being processed. Both the water sample and the soil sample were analyzed for their chemical and microbial characteristics.
Figure 1.
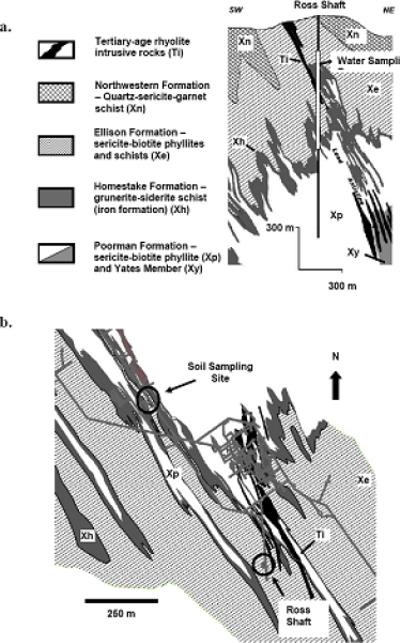
a. Cross section of the Homestake underground mine showing the location of the water sampling along the extent of the Ross Shaft (244 – 914 m). b. Plan view of the 2000 level (610 m below the surface) showing the location of the soil sampling site in relation to the Ross Shaft and the geologic map of that level.
Chemical analysis
Chemical components of all samples used in this study were analyzed by Midcontinent Testing Laboratories, Inc. (Rapid City, SD), a U. S. Environmental Protection Agency (EPA)–certified facility, in accordance with standard EPA procedures. Briefly, soil samples were suspended in deionized water, stored at room temperature for 24 hours, and centrifuged. The supernatant was used for all further analyses, except the metal concentration determination. Table 1 lists geochemical components of the DUSEL samples and analysis methods used for this study. Cation concentrations were measured with the AA400 atomic absorption spectrometer (Varian, Inc) and anions by the EPA–guided specific methods. For determination of metal concentrations, 2 g of soil sample was dissolved in 10 mL of concentrated nitric acid (EPA 3051A) and 50 mL of water sample was digested in 5 mL of concentrated nitric acid, evaporated down to 5 mL and reconstituted to the volume specified by EPA 3030E. The digested product from each sample was determined using inductively coupled plasma mass spectrometry (ICP – MS) in an ELAN® 9000 DRC™ (Perkin Elmer).
Table 1.
Geochemical compositions of DUSEL samples.
| Parameter | Water1 (244 – 914 m) 5/25/2007 | Soil2 (610 m) 9/11/2007 | Analysis Method |
|---|---|---|---|
| pH | 8.00 (mg/L) | 7.49 (mg/kg) | EPA SW846 045C |
| TIC | ND | 26.6 | 2SM 5310 C |
| TOC | ND | <5.00 | 2SM 5310 C |
| TDS | 2,230 | ND | 1EPA 160.1 |
| Anions | |||
| Chloride (Cl–) | 25 | 1.25 | SM 4500-Cl B |
| Nitrite (NO2–) | <0.050 | 0.107 | 1EPA 353.2 |
| 2SM 4500-NO2 B | |||
| Nitrate (NO3–) | 0.203 | 11.0 | 1EPA 353.2 |
| 2SM 4500-NO3 B | |||
| Sulfate (SO42–) | 1,580 | 6,000 | EPA 375.2 |
| Phosphate (PO43–) | ND | <0.050 | SM 4500-P E |
| Thiocyanate (SCN–) | 0.116 | <0.500 | ASTM D4193 |
| Cations | |||
| Ammonia (NH4+) | 0.143 | 4.75 | 1EPA 350.3 |
| 2SM 4500-NH3 B | |||
| Calcium (Ca2+) | 250 | 476 | SM 3111 B |
| Magnesium (Mg2+) | 267 | 190 | SM 3111 B |
| Manganese (Mn2+) | 0.334 | 1,850 | EPA 200.8 |
| Potassium (K+) | 30.9 | 57.4 | SM 3111 B |
| Sodium (Na+) | 101 | 24.9 | SM 3111 B |
| Metals | |||
| Aluminum (Al) | 0.03 | 13,500 | EPA 200.8 |
| Arsenic (As) | 0.016 | 1,520 | EPA 200.8 |
| Barium (Ba) | 0.037 | 30.0 | EPA 200.8 |
| Chromium (Cr) | <0.001 | 10.5 | 1EPA 200.8 |
| 2EPA 200.8 DRC | |||
| Copper (Cu) | <0.005 | 49.9 | EPA 200.8 |
| Iron (Fe) | 1.86 | 78,800 | EPA 200.8 |
| Gold (Au) | <0.001 | <0.01 | EPA 231.2 |
| Lead (Pb) | <0.001 | 9.19 | EPA 200.8 |
| Molybdenum (Mo) | 0.003 | 0.265 | EPA 200.8 |
| Nickel (Ni) | 0.005 | 7.85 | EPA 200.8 |
| Selenium (Se) | <0.005 | 1.57 | EPA 200.8 |
| Silver (Ag) | <0.001 | <0.200 | EPA 200.8 |
| Zinc (Zn) | <0.050 | 31.4 | EPA 200.8 |
TIC: Total Inorganic Carbon TOC: Total Organic Carbon
TDS: Total Dissolved Solids ND: Not Determined
SM: Standard Methods ASTM: American Society for Testing and Materials
Molecular analysis of microbial communities
DNA was extracted from the cells collected by centrifugation of 3 L of the water sample and from 500 mg of soil sample using the Fast DNA® Soil Extraction Kit (Qbiogene). Concentrations of DNA were determined using a NanoDrop® ND–1000 spectrophotometer (Thermo Scientific). The polymerase chain reaction (PCR) was used to amplify the 16S rDNA gene from the extracted DNA for use in the construction of bacterial and archaeal clone libraries. The bacterial and archaeal PCRs were performed on an iCycler® thermal cycler (BioRad) and consisted of approximately 50 ng of sample DNA, 25 μL of 2× Master Mix (Promega), 0.5 μM of forward primer (530F [5’–GTGCCAGCMGCCGCGG–3’]) and reverse primer (1490R [5’–GGTTACCTTGTTACGACTT–3’]) for Bacteria [24], or 0.25 μM of forward primer (21F [5’–TTCCGGTTGATCCTGCCGGA–3’]) [25] and reverse primer (1492R [5’–GGTTACCTTGTTACGACTT–3’] [24] for Archaea, and DNase free water for a total volume of 50 μL. Thermal cycling parameters were as follows: Initial denaturation at 94°C for 4 min; 30 cycles of 94°C for 30 sec, 56°C for 1 min, and 72°C for 2 min; followed by incubation at 72°C for 15 min; and cooling to 4°C prior to removal. After examination of PCR products for size by agarose gel electrophoresis, they were excised from the gel and cleaned using the Wizard® SV Gel and PCR Clean–up System (Promega) prior to ligation. Clone libraries were constructed using the pGEM®T–Easy Vector System II with JM109 competent cells (Promega). Sequencing reactions were done using M13 (–20) forward and M13 (–27) reverse primers with a BigDye® Terminator v3.1 Ready Reaction Cycle Sequencing kit (Applied Biosystems) on an ABI 3130 Genetic Analyzer (Applied Biosystems). Chromatograms were edited and contiguous sequences assembled using Sequencher® v4.8 (GeneCodes).
Phylogenetic analysis of sequences
Contiguous sequences were screened for putative chimeras using the Bellerophon program [26], aligned using MEGA4 software [27], and loaded into the DNADIST function of PHYLIP [28] to generate Jukes–Cantor corrected distance matrices [29]. Distance matrices were inputted into DOTUR [30] and the furthest–neighbor method was used to determine the operational taxonomic units (OTUs) based on 97% raw sequence similarity. Bacterial sequences were aligned against the SILVA [31] and Greengenes [32] 16S ribosomal DNA databases, as well as inputted into the SeqMatch tool of the Ribosomal Database Project (RDP) II [33] to obtain the nearest related sequences for use as references in the phylogenetic trees. The same databases were used for archaeal sequence sets replacing RDPII with the BLASTN search program of the National Center for Biotechnology Information (NCBI). Using MEGA4 [27], all trees were created by the neighbor–joining method to infer evolutionary history [34] and the significance levels of interior branch points obtained were determined using the bootstrap test for phylogeny (10,000 data resamplings) [35].
Non–parametric species richness estimates were made using the Chao1 bias–corrected richness estimator [36,37] at a genetic distance of 0.03 in DOTUR. Additionally, DOTUR was used to calculate statistical parameters including the Simpson diversity index (D), the Shannon diversity index (H′) with 95% confidence intervals (CI), and rarefaction analysis for comparison of DUSEL community diversities. The estimated steepness values (angle θ) of the line tangent to the rarefaction curves generated by DOTUR were also determined as an indication of the ability of the sampling effort to reflect the fullness of the diversity within each sample. The software program SONS [38] was used to estimate the similarity between the bacterial water and soil communities, and between the archaeal water and soil communities. The Libshuff/S-Libshuff function of MOTHUR v1.8.0 [39] was used to test whether or not communities shared the same structure. After Bonferoni correction, the critical P-value at which two libraries were considered significantly different in community composition was p ≤0.025. AMOVA analysis was done in Arlequin v3.11 [40] to test whether or not the genetic diversity between the communities was different.
Nucleotide sequence accession numbers
The 16S rDNA sequences from a total of 216 DUSEL clone isolates were submitted to GenBank and assigned under accession numbers FJ718772 – FJ718905 for Bacteria members and FJ718906 – FJ718987 for Archaea members.
Results
Geochemistry of the DUSEL samples
The geochemical properties of the water and soil samples are summarized in Table 1. The water and soil samples are both alkaline (pH 8.00 and 7.49, respectively). The soil sample contained significantly high concentrations of metals such as Al, As, Ba, Cu, Fe, Zn, Cr, Pb, and Ni. Chlorinity was considerably higher in the water than in the soil, but the concentrations of metals and most other cations and anions in water were notably lower than those in soil. The available carbon in the soil was mainly inorganic with less than 5mg/kg of organic carbon present. Since the gold deposits at the Homestake mine are typically associated with the iron formation, large amounts of Fe were expected in both types of DUSEL samples [3]. Nonetheless, compared to the soil sample that showed a total concentration of Fe at 78,800 mg/kg, the water sample contained 1.86 mg/L of total Fe and less than 0.050 mg/L of dissolved Fe. The water contained 2,230 mg/L of total dissolved solids. High sulfate concentrations were found in both the water (1,580 mg/L) and soil (6,000 mg/kg) samples.
Composition of microbial communities
Archaeal Communities
After removal of chimeric sequences from the dataset, 21 clones of archaeal 16S rDNA sequences remained in the water sample and 61 in the soil sample. At 97% sequence similarity DOTUR identified three OTUs (OTUs 1A–1, 1A–2, and 1A–3) from the water sample, and one OTU (OTU 2A–1) from the soil sample. BLAST and phylogenetic analyses of the 16S rDNA gene indicated that both of the archaeal communities were comprised of OTUs most closely related to members of the newly proposed phylum Thaumarchaeota [41]. Based on their position in the 16S rDNA tree OTUs 1A–1, 1A–3 and 2A–1 were most closely related to uncultured members of the Thaumarchaeota (Figure 2). In the phylogenetic tree, OTU 1A–3 groups with members of the Cenarchales, while OTU 1A–1 and 2A–1 grouped as a sister clade to members of the Nitrosopumilales. BLAST analysis of the near full–length 16S rDNA sequence confirmed that OTU 1A–1 and OTU 2A–1 were 96% identical to Nitrosopumilus maritimus (NC010085) with 100% query coverage, such that these OTUs would be confidently placed in the genus Nitrosopumilus, but may represent a novel species. Similarly, BLAST analysis of OTU 1A–3 supported its placement in the tree with 100% query coverage and 95% identity to uncultured marine Thaumarchaeota. However, OTU 1A–2 was a unique phylotype within the water Archaea dataset, BLAST analysis of which returned a few hits that showed 99% query coverage with 74–75% maximum identity to uncultured Archaea sequences isolated from environmental samples. Interestingly, there were no Crenarchaeota members identified from the two samples used in this study, although a previous study [10] indicated the presence of Crenarchaeota from DUSEL soil samples from a similar environment 700 m deeper than the sampling site for this study.
Figure 2.
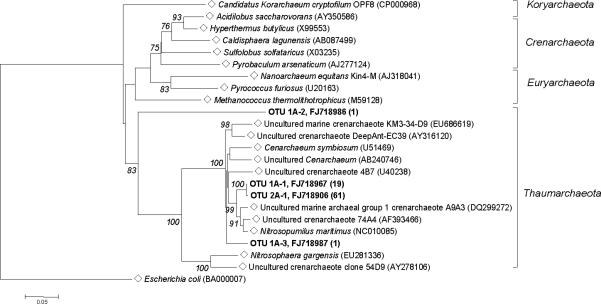
Phylogenetic tree showing the relationship of 16S rDNA sequences of Homestake Archaea OTUs from water (1A-1, 1A-2 and 1A-3) and soil (2A-1) with reference sequences (◇) selected from GenBank and Greengenes. Bootstrap values (10,000 data-resampling) above 75% are shown. Clones sequenced are in bold, showing the OTU and representative GenBank accession number with the number of members in parenthesis. The scale bar represents 0.05 substitutions per nucleotide position. Tree is rooted with Escherichia coli (BA000007) as an outgroup.
Bacterial communities
After removal of chimeric sequences from the dataset, 28 clones of bacterial 16S rDNA sequences from the water sample and 106 clones from the soil sample remained. DOTUR identified 21 OTUs in the bacterial water sample, and 27 OTUs in the bacterial soil sample. Bacterial communities in the DUSEL water sample were comprised largely of phylum Proteobacteria (79%), within which the sub–groups included Alphaproteobacteria (32%), Betaproteobacteria (41%), and Gammaproteobacteria (27%) (Figure 3). Additional phyla represented at a lower abundance in Bacteria were identified as Bacteroidetes (7%), Planctomycetes (3.5%), Verrucomicrobia (7%), and Firmicutes (3.5%). Within the Betaproteobacteria, all nine of the sequences are highly similar to members of the order Burkholderiales. Two sequences, OTU 1B–5 and OTU 1B–12, showed 92% and 77% identity respectively to members of the Gammaproteobacteria class based on RDP II classifier results, indicating these sequences may represent a novel order. OTU 1B–9 represented a novel Firmicute phylotype based on 100% query coverage with only 86% identity to members of the family Planococcaceae.
Figure 3.

Phylogenetic tree showing the relationship of 16S rDNA sequences from Homestake water Bacteria OTUs with reference sequences (◇) obtained from GenBank. Bootstrap values (10,000 data re-samplings) above 75% are shown. Clones sequenced are in bold, showing the OTU and representative GenBank accession number with the number of members in parenthesis. The scale bar represents 0.05 substitutions per nucleotide position. Tree is rooted with Methanococcus thermolithotrophicus (M59128) as an outgroup.
The majority of the DUSEL soil Bacteria were assigned to the phylum Proteobacteria (95%), 70% of which were comprised of Gammaproteobacteria (Figure 4). Among the Gammaproteobacteria sequences, approximately 43.6% showed 99% to 100% query coverage and 91% to 92% maximum identity to sequences from several uncultured Gammaproteobacteria from environments such as deep sea hydrothermal vents, sea sediments, and hypersaline lakes. The nearest cultured relatives include the extremophilic bacterium Thiohalomonas nitratireducens (DQ836238). Forty-eight percent (48%) were identified as being members of the order Xanthomonadales, and approximately 2.8% were classified as members of the orders Chromatiales, Alteromonadales, or Thiotrichales. The remaining 30% of Proteobacteria was comprised of Alphaproteobacteria (13%), Betaproteobacteria (15%), and Deltaproteobacteria (2%). The soil community also included the phyla Bacteroidetes (2%), Firmicutes (2%), and Actinobacteria (1%). Although Betaproteobacteria from both samples were classified as belonging to the order Burkholderiales, no individual members are identical to one another. Among the DUSEL Proteobacteria, the Gammaproteobacteria class was much more prevalent in the soil ecosystem, whereas the Betaproteobacteria class was more prevalent in the water.
Figure 4.

Phylogenetic tree showing the relationship of 16S rDNA sequences from Homestake soil Bacteria OTUs with reference sequences (◇) obtained from GenBank. Bootstrap values (10,000 data re-samplings) above 75% are shown. Clones sequenced are in bold, showing the OTU and representative GenBank accession number with the number of members in parenthesis. The scale bar represents 0.05 substitutions per nucleotide position. Tree is rooted with Methanococcus thermolithotrophicus (M59128) as an outgroup.
Statistical comparison of community diversities
The Chao1 bias–corrected coverage estimates were 32% and 73% for the water and soil Bacteria, respectively, and 75% and 100% for the water and soil Archaea, respectively (Table 2). Rarefaction analyses of DUSEL 16S rDNA sequences generated significantly different curves for the two samples (Figure 5), in which the steepness values (θ) are in agreement with the Chao1 coverage estimates. Statistical analyses of microbial phylotype diversities in environments are often problematic, mainly because exhaustive sampling and identification of every member of the community are difficult to accomplish [42]. Nonetheless, greater diversity was found among the Bacteria than among the Archaea in both the soil and water samples.
Table 2.
Diversity indices based on analysis with DOTUR.
| # OTUs Observed | Chao1# OTUs Predicted | Chao1 95% CI | Chao1 % Coverage | Simpson Index (D) | Shannon Index (H′) | Pielou's Evenness Index (E) | |
|---|---|---|---|---|---|---|---|
| Archaea | |||||||
| Water | 3 | 4 | 3.08 –15.92 | 75 | 0.814 | 0.38 ± 0.39 | 0.35 |
| Soil | 1 | 1 | 1 | 100 | 1 | 0 ± 0.02 | NA |
| Bacteria | |||||||
| Water | 21 | 66.333 | 34.09 – 178.03 | 31.66 | 0.029 | 2.92 ± 0.30 | 0.96 |
| Soil | 27 | 36.750 | 29.67 – 62.54 | 73.47 | 0.106 | 2.63 ± 0.22 | 0.80 |
NA: Not Available
Figure 5.
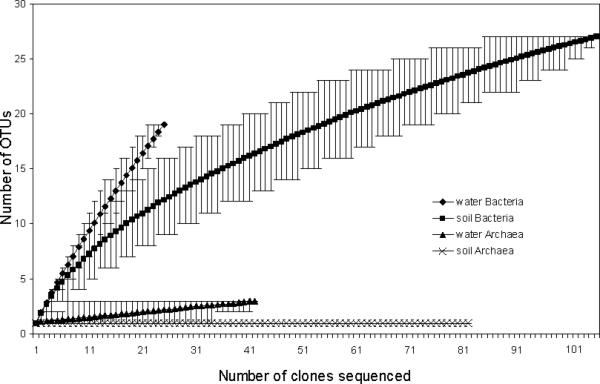
Rarefaction curves of 16S rDNA sequences of Bacteria and Archaea clone isolates from DUSEL water and soil samples at the species level (distance ≤ 0.03). The steepness value (angle θ) was obtained from the estimated tangent line to the rarefaction curve by using its terminal two points: water Bacteria θ = 30.92°; soil Bacteria θ = 6.90°; water Archaea θ = 5.71°; and soil Archaea θ = 0°.
Diversity indices revealed large differences in species richness between the bacterial and archaeal communities. Values of Simpson's index (D) (Table 2) for the bacterial communities were 0.03 and 0.11 from the water and soil samples, respectively, whereas those for the archaeal communities were 0.81 and 1.00 from the water and soil samples, respectively. Shannon diversity indices (H′) for the soil (H′ = 2.92) and the water (H′ = 2.63) Bacteria as well as for the soil (H′ = 0.38) and the water (H′ = 0) Archaea are in agreement with the data of Simpson's diversity analyses. Pielou's [43] evenness index (E) was estimated to assess the evenness of species representation within each community. Bacteria water and soil communities with E values of 0.96 and 0.80, respectively, appeared to have a moderately uniform level of species evenness. The evenness score for the Archaea water sample (E = 0.35) numerically depicted the skew in species evenness evident in the phylogenetic tree where 95% of the species were classified as belonging to a single unidentified species of Thaumarchaeota. These statistical analyses confirmed the notion that the Bacteria in both DUSEL environments were highly diverse compared to the Archaea in both environments and, furthermore, that the water community contained a higher level of microbial diversity than the soil community. In addition, SONS sequence similarity analysis estimated that there were no shared OTUs at the species or genus level (genetic distance of 0.03 to 0.07) between the two samples neither within the bacterial communities nor within the archaeal communities. S-Libshuff analysis of the bacterial communities indicated the water and soil communities did not share the same structure, that is they differed in community composition, (soil – water comparison, p ≤0.0001; water – soil comparison, p = 0.0004). Good's coverage estimates for soil and water bacteria were 0.85 and 0.39, respectively. AMOVA analysis of the bacterial communities also indicated the two communities were significantly different (FST = 0.103, p <0.0001). S-Libshuff analysis of the archaeal communities was confounded by the fact that the sampling of the soil and water Archaea were sufficiently high that Good's coverage estimate was 1.00 and 0.96, respectively, at a distance of only 0.03 making the homologous and heterologous coverage curves similar. However, AMOVA analysis of the water and soil Archaea communities did indicate that the two communities are significantly different (FST = 0.0516, p <0.0001).
Discussion
The phylogenetic diversity throughout DUSEL microbial communities hinges not only upon geochemical attributes of the various rock formations, but also upon geochemical alterations induced by mining. The geochemistry of the 610 m soil sample reflects the representative characteristics of the Homestake Formation, which is mainly comprised of carbonate– and silica–dominated rocks rich in iron, manganese, magnesium, and sulfur, but poor in phosphate [3]. Soluble carbonates likely maintain a higher pH by serving as a buffer, although a locally lower pH can be expected where water is in contact with pyrite in the Tertiary igneous rocks of the Homestake Formation. Carbon dioxide may serve as a carbon source for chemolithoautotrophs, in which CO2 is fixed by scavenging electrons generated via oxidation of iron, sulfur, and manganese. Further studies are necessary to explore the relationship between microbial populations and geochemical components in DUSEL soil and water habitats. However, the taxa whose 16S rDNA sequences were found to be present in the communities can provide clues to the diversity of metabolic pathways present within the communities occupying drift wall surfaces, water seeps and soils found in the DUSEL subsurface. This is particularly true in the case of the Archaea 16S sequences identified from both the water and soil samples.
The geochemical properties described herein were consistent with the presence of soil and water Archaea represented by clones of OTU 2A–1 and 1A–1, respectively, which grouped together as a sister clade to N. maritimus, an aerobic mesophilic, carbon–fixing, ammonia–oxidizing chemoautotroph of the phylum Thaumarchaeota [41,44,45]. Similarly, one of the OTUs, 1A–3, was grouped with an uncultured Cenarchaeum most closely related to a marine sponge endosymbiont, Cenarchaeum symbiosum, of the phylum Thaumarchaeota. Both N. maritimus and C. symbiosum harbor genes for ammonia oxidation [45,46]. The dominance of mesophilic ammonia oxidizing Archaea in soils [47] and marine environments [48-50] have been recently recognized. As further exploration of microbial diversity continues at deeper, pristine regions of the DUSEL at Homestake, a survey of the genes involved in ammonia oxidation pathways will shed light on the mechanism and extent of biogeochemical cycling of nitrogen in the deep subsurface and the role that Archaea play in that process. The presence of Thaumarchaeota at this shallower level of the DUSEL subsurface is contrasted with the findings of Rastogi et al. [10] in which, at 1.34 km below the surface in the Homestake DUSEL, the majority of the members of the soil archaeal community were identified as members of the class Thermoprotei of Crenarchaeota. Furthermore, investigation of archaeal communities of subsurface fluids from a South African mine [17] identified that the mine water contained predominantly Crenarchaeota, whereas borehole fluids were populated with mainly Euryarchaeota.
The majority of bacterial groups identified from the DUSEL water and soil samples belonged to the Proteobacteria. Members of this phylum are widely distributed in surface soils and waters [51,52]. The prevalence of Betaproteobacteria was much greater in the DUSEL water compared to the soil and has also been reported to be ubiquitous in global aquatic systems from freshwater ecosystems of the northern hemisphere [53] to boreholes of a mine in South Africa [54]. Although Gammaproteobactera are often found in soil and silty environments, their unusually high abundance (70%) relative to other proteobacterial subgroups in soil appears somewhat unique to the DUSEL community. The presence of bacterial members that are common in surface environments is not surprising because the DUSEL samples were taken less than 1 km below ground, a relatively shallow subsurface. It is of particular interest to note, however, that despite good coverage of the soil bacteria library, members of the DUSEL Bacteria from both water and soil samples appeared to be diverse and independent of each other's community (Figures 3 and 4). Perhaps this is driven by geochemical differences between the two subsurface niches causing differential survival of both native and introduced taxa. However, it is also possible that increased coverage of the water bacterial library might reveal shared taxa.
The most prevalent anion in the soil sample was sulfate (6,000 mg/kg). It was not surprising then that 46% percent of the soil bacteria 16S rDNA sequences (OTUs 2B–3, 2B–8, 2B–9, 2B–11, and 2B–14) were identified as members of phyla capable of metabolizing sulfur. For example, soil OTU 2B–8 (14 members) was classified by the RDP II as being closely related (96% identical) to Thiobacillus plumbophilus. This chemolithoautotrophic species is capable of oxidizing H2S and galena (PbS), but not iron [55]. Similarly, soil OTU 2B–3 (26 members) grouped to the same order (92% similarity) as Thiohalomonas nitratireducens. This species is an obligate chemolithoautotroph that utilizes thiosulfate as an electron donor to yield sulfate as the final oxidation product [56].
Based on 16S rDNA sequences, our findings indicated the presence of OTUs that grouped closely to several genera that contain species with unique metabolic capabilities. Interesting taxa identified in the water sample included OTU 1B–1 with 2 members (7.1%) that grouped to the order (>88% identical) Verrucomicrobiales. Members of the phylum Verrucomicrobia are common to a variety of aquatic systems including marine sediments and hot springs [57,58]. In the soil sample, OTU 2B–27 grouped with Alcaligenes spp. (97% identical). Members of this genus include the facultative chemolithoautotrophic hydrogen bacterium, A. eutrophus [59], more recently known as Ralstonia metallidurans that was isolated from heavy metal–contaminated environments [60]. The metabolic capabilities associated with these genera are consistent with the geochemistry of the drift soil from which the isolates were taken. Furthermore, this OTU 2B–27 grouped as a sister clade to Paucimonas lemoignei, formerly known as Pseudomonas lemoignei. Mudder and Whitlock [61] developed a cyanide–degrading mutant strain, Pseudomonas paucimobilis mudlock ATCC 39204, from P. paucimobilis that was isolated from the Homestake wastewater treatment effluent by acclimating to high concentrations of cyanide, metal cyanide complexes, and thiocyanates. The unique metabolic pathway harbored by this species has been used for over a decade in the treatment of Homestake wastewater effluent and illustrates the potential utility that may be offered by other novel DUSEL microbes. Similarly, OTU 2B–14 with two clones was identified as 98% identical to Halothiobacillus species of the order Chromatiales, whose members are known to be capable of chemoautotrophic growth by oxidizing sulfur, sulfide or thiosulfate in the presence of oxygen as the electron acceptor [62]. It is interesting to note that while many members of the Chromatiales are phototrophic purple sulfur bacteria, members of the genus Halothiobacillus are not photosynthetic. Therefore, the subsurface ecological niche from which these sequences were found would be consistent with the metabolic capabilities of the genus. There was one soil OTU identified as being 96-97% identical to members of the genus Lysobacter (9% of the clones). Members of the genus Lysobacter have been isolated from soils globally and are known to produce enzymes with biotechnological potential as well as novel pharmaceutically valuable antibiotics [63-65]. Although a significantly high content of iron was detected in the 610 m soil sample, there were no sequences identified as belonging to bacterial taxa with iron–metabolizing capabilities. This is in contrast to a recent study in which the microbial diversity of soil at 1.34 km below the surface of the Homestake DUSEL was characterized and found to harbor sequences that grouped closely to bacteria such as Acidithiobacillus ferooxidans, an iron–oxidizing bacterium [10]. Both the Chromatiales and the Thiotrichales orders contain chemolithoautotrophic species capable of growing in a variety of mesic and extreme environments [66-70]. Interesting phylotypes found in the DUSEL soil were identified as Rhodanobacter (98% identical), Dyella (97% identical), and Lysobacter (96-97% identical), many of which are closely related to unknown and uncultured strains isolated from soil and lake sediments [71-73]. Recently, cellulolytic mesophiles and thermophiles were identified from the Homestake DUSEL [9]. Unique biochemical characteristics of cellulases and hemicellulases from the Homestake thermophiles are currently being investigated in our laboratories.
The geochemistry of these subsurface environments has shaped unique niches that can be exploited by extremophiles whether exogenous or endogenous. The co–occurrence of marine taxa with terrestrial and freshwater taxa also suggests that localized microniches play a significant role in shaping the biodiversity and the overall microbial metabolic activities of deep subsurface ecosystems. It can be hypothesized that at the borders of these microniches horizontal gene transfer events may be more apt to occur and could give rise to unique niche specific combinations of metabolic activities [74]. Further investigation of microbial community structures over a wide collection of DUSEL samples will contribute to a better understanding of the relative distributions and origins of inhabitants and their phylogenetic lineages within DUSEL extreme environments. Such comprehensive information will also allow us to measure and understand the significance of microbial diversity and their physiologic variability throughout the Homestake DUSEL geosphere as well as the impact of introduced organisms on the ecology and evolution of native deep subsurface extremophile communities. Therefore, bioprospecting for extremophiles with novel metabolic activities will be particularly fruitful at this former Homestake gold mine.
Acknowledgements
The authors are thankful to John Schneider of the Department of Geology/Geological Engineering, South Dakota School of Mines and Technology, for critical information on the Homestake geology. Special thanks go to the South Dakota Science and Technology Authority for providing the DUSEL samples and their geochemical data. This work was funded in part by the SD NASA–EPSCoR Program (NASA Grant#: NCC5–588), by the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) grant number 2P20RR016479, and by a partial match from the Western South Dakota DNA Core Facility (WestCore) at Black Hills State University, Spearfish, SD.
References
- 1.Horneck G. Exobiology, the study of the origin, evolution and distribution of life within the context of cosmic evolution: a review. Planet Space Sci. 1995;43:189–217. doi: 10.1016/0032-0633(94)00190-3. [DOI] [PubMed] [Google Scholar]
- 2.Newman DK, Banfield JF. Geomicrobiology: how molecular-scale interactions underpin biogeochemical systems. Science. 2002;296:1071–1077. doi: 10.1126/science.1010716. [DOI] [PubMed] [Google Scholar]
- 3.Caddey SW, Bachman RL, Campbell TJ, Reid RR, Otto RP. The Homestake gold mine, an early Paleozoic iron–formation–hosted gold deposit, Lawrence County, South Dakota. In: Shawe DR, Ashley RP, Carter LMH, editors. Geology and resources of gold in the United States. 1857-J. U.S. Geol. Surv. Bull.; 1991. pp. J1–J67. [Google Scholar]
- 4.Duke GI. Ph.D. South Dakota School of Mines and Technology; 2005. Geochemistry and geochronology of Paleocene–Eocen alkalic igneous rocks, northern Black Hills, South Dakota and Wyoming. [Google Scholar]
- 5.Bachman RL, Caddey SW. The Homestake mine, Lead, South Dakota: An overview. In: Patersonand CJ, Lisenbee AL, editors. Metallogeny of gold in the Black Hills, South Dakota. Guidebook Prepared for Soc. Econ. Geol. Field Conf.; 1990. pp. 89–94. [Google Scholar]
- 6.Mathisrud GD, Sumner JS. Underground induced polarization surveying at the Homestake mine. Min. Congr. J. 1967;53:66–69. [Google Scholar]
- 7.Meier LF. Structure and ore trend description of the Homestake mine. In: Patersonand CJ, Lisenbee AL, editors. Metallogeny of gold in the Black Hills, South Dakota. Guidebook Prepared for Soc. Econ. Geol. Field Conf.; 1990. pp. 103–111. [Google Scholar]
- 8.Rye DM, Rye RO. Homestake gold mine, South Dakota: I. Stable isotope studies. Econ. Geol. 1974;69:293–316. [Google Scholar]
- 9.Rastogi G, Muppidi GL, Gurram RN, Adhikari A, Bischoff KM, Hughes SR, Apel WA, Bang SS, Dixon DJ, Sani RK. Isolation and characterization of cellulose-degrading bacteria from the deep subsurface of the Homestake gold mine, Lead, South Dakota, USA. J. Ind. Microbiol. Biotechnol. 2009;36:585–598. doi: 10.1007/s10295-009-0528-9. [DOI] [PubMed] [Google Scholar]
- 10.Rastogi G, Stetler LD, Peyton BM, Sani RK. Molecular analysis of prokaryotic diversity in the deep subsurface of the former Homestake gold mine, South Dakota, USA. J. Microbiol. 2009;47:371–384. doi: 10.1007/s12275-008-0249-1. [DOI] [PubMed] [Google Scholar]
- 11.Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez RJ, Wang Y, Raimondo MA, Coombs JM, Barkay T, Sobecky PA. Horizontal gene transfer of PIB-type ATPases among bacteria isolated from radionuclide- and metal-contaminated subsurface soils. Appl. Environ. Microbiol. 2006;72:3111–3118. doi: 10.1128/AEM.72.5.3111-3118.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaksonen AH, Plumb JJ, Robertson WJ, Spring S, Schumann P, Franzmann PD, Puhakka JA. Novel thermophilic sulfate-reducing bacteria from a geothermally active underground mine in Japan. Appl. Environ. Microbiol. 2006;72:3759–3762. doi: 10.1128/AEM.72.5.3759-3762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deflaun MF, Fredrickson JK, Dong H, Pfiffner SM, Onstott TC, Balkwill DL, Streger SH, Stackebrandt E, Knoessen S, van Heerden E. Isolation and characterization of a Geobacillus thermoleovorans strain from an ultra-deep South African gold mine. Syst. Appl. Microbiol. 2007;30:152–164. doi: 10.1016/j.syapm.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM, Saar MO, Alexander S, Alexander EC, Jr., Rohwer F. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gihring TM, Moser DP, Lin L-H, Davison M, Onstott TC, Morgan L, Milleson M, Kieft TL, Trimarco E, Balkwil DL, Dollhopf ME. The distribution of microbial taxa in the subsurface water of the Kalahari Shield, South Africa. Geomicrobiol. J. 2006;23:415–430. [Google Scholar]
- 18.Takai K, Moser DP, DeFlaun M, Onstott TC, Fredrickson JK. Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 2001;67:5750–5760. doi: 10.1128/AEM.67.21.5750-5760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haveman SA, Pedersen K, Ruotsalainen P. Distribution and metabolic diversity of microorganisms in deep igneous rock aquifers of Finland. Geomicrobiol. J. 1999;16:277–294. [Google Scholar]
- 20.Sahl JW, Schmidt R, Swanner ED, Mandernack KW, Templeton AS, Kieft TL, Smith RL, Sanford WE, Callaghan RL, Mitton JB, Spear JR. Subsurface microbial diversity in deep-granitic-fracture water in Colorado. Appl. Environ. Microbiol. 2008;74:143–152. doi: 10.1128/AEM.01133-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fredrickson JK, Balkwill DL. Geomicrobial processes and biodiversity in the deep terrestrial subsurface. Geomicrobiol. J. 2006;23:345–356. [Google Scholar]
- 22.Lin LH, Slater GF, Lollar BS, Lacrampe–Coulloume G, Onstott TC. The yield and isotopic composition of radiolytic H2, a potential energy source for the deep subsurface biosphere. Geochim. Cosmochim. Acta. 2005;69:893–903. [Google Scholar]
- 23.Onstott TC, Moser DP, Pfiffner SM, Fredrickson JK, Brockman FJ, Phelps TJ, White DC, Peacock A, Balkwill D, Hoover R, Krumholz LR, Borscik M, Kieft TL, Wilson R. Indigenous and contaminant microbes in ultradeep mines. Environ. Microbiol. 2003;5:1168–1191. doi: 10.1046/j.1462-2920.2003.00512.x. [DOI] [PubMed] [Google Scholar]
- 24.Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandtand E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematic. John Wiley and Sons; New York: 1991. pp. 115–175. [Google Scholar]
- 25.DeLong EF. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 1992;89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–2319. doi: 10.1093/bioinformatics/bth226. [DOI] [PubMed] [Google Scholar]
- 27.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 28.Felsenstein J. PHYLIP – Phylogeny inference package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 29.Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism, in Mammalian protein metabolism. Academic Press; New York: 1969. pp. 21–132. [Google Scholar]
- 30.Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 35.Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 36.Chao A. Non–parametric estimation of the number of classes in a population. Scand. J. Stat. 11. 1984:265–270. [Google Scholar]
- 37.Chao A, Ma MC, Yang MCK. Stopping rules and estimation for recapture debugging with unequal failure rates. Biometrika. 1993;80:193–201. [Google Scholar]
- 38.Schloss PD, Handelsman J. Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl. Environ. Microbiol. 2006;72:6773–6779. doi: 10.1128/AEM.00474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 41.Brochier–Armanet C, Boussau B, Gribaldo S, Forterre P. Mesophilic crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 2008;6:245–252. doi: 10.1038/nrmicro1852. [DOI] [PubMed] [Google Scholar]
- 42.Schloss PD, Handelsman J. Toward a census of bacteria in soil. PLoS Comput. Biol. 2006;2:e92. doi: 10.1371/journal.pcbi.0020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pielou EC. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966;13:131–144. [Google Scholar]
- 44.You J, Das A, Dolan EM, Hu Z. Ammonia-oxidizing archaea involved in nitrogen removal. Water Res. 2009;43:1801–1809. doi: 10.1016/j.watres.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 45.Konneke M, Bernhard AE, de la Torre JR, Walker CB, Waterbury JB, Stahl DA. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature. 2005;437:543–546. doi: 10.1038/nature03911. [DOI] [PubMed] [Google Scholar]
- 46.Ingalls AE, Shah SR, Hansman RL, Aluwihare LI, Santos GM, Druffel ER, Pearson A. Quantifying archaeal community autotrophy in the mesopelagic ocean using natural radiocarbon. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6442–6447. doi: 10.1073/pnas.0510157103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, Prosser JI, Schuster SC, Schleper C. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- 48.Mincer TJ, Church MJ, Taylor LT, Preston C, Karl DM, DeLong EF. Quantitative distribution of presumptive archaeal and bacterial nitrifiers in Monterey Bay and the North Pacific Subtropical Gyre. Environ. Microbiol. 2007;9:1162–1175. doi: 10.1111/j.1462-2920.2007.01239.x. [DOI] [PubMed] [Google Scholar]
- 49.Wuchter C, Abbas B, Coolen MJ, Herfort L, van Bleijswijk J, Timmers P, Strous M, Teira E, Herndl GJ, Middelburg JJ, Schouten S, Sinninghe Damste JS. Archaeal nitrification in the ocean. Proc. Natl. Acad. Sci. U. S. A. 2006;103:12317–12322. doi: 10.1073/pnas.0600756103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam P, Jensen MM, Lavik G, McGinnis DF, Muller B, Schubert CJ, Amann R, Thamdrup B, Kuypers MM. Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc. Natl. Acad. Sci. U .S. A. 2007;104:7104–7109. doi: 10.1073/pnas.0611081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hengstmann U, Chin KJ, Janssen PH, Liesack W. Comparative phylogenetic assignment of environmental sequences of genes encoding 16S rRNA and numerically abundant culturable bacteria from an anoxic rice paddy soil. Appl. Environ. Microbiol. 1999;65:5050–5058. doi: 10.1128/aem.65.11.5050-5058.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Sullivan LA, Weightman AJ, Fry JC. New degenerate Cytophaga-Flexibacter-Bacteroides-specific 16S ribosomal DNA-targeted oligonucleotide probes reveal high bacterial diversity in River Taff epilithon. Appl. Environ. Microbiol. 2002;68:201–210. doi: 10.1128/AEM.68.1.201-210.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zwart G, Crump BC, Agterveld M.P.K.-v., Hagen F, Han SK. Typical freshwater bacteria: An analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 2002;28:141–155. [Google Scholar]
- 54.Lin L-H, Gihring TM, Lollar BS, Boice E, Pratt LM, Lippmann-Pipke J, Bellamy RES, Hall JA, Onstott TC. Planktonic microbial communities associated with fracture-derived groundwater in a deep gold mine of South Africa. Geomicrobiol. J. 2006;23:475–497. [Google Scholar]
- 55.Drobner E, Huber H, Rachel R, Stetter KO. Thiobacillus plumbophilus spec. nov., a novel galena and hydrogen oxidizer. Arch. Microbiol. 1992;157:213–217. doi: 10.1007/BF00245152. [DOI] [PubMed] [Google Scholar]
- 56.Sorokin DY, Tourova TP, Braker G, Muyzer G. Thiohalomonas denitrificans gen. nov., sp. nov. and Thiohalomonas nitratireducens sp. nov., novel obligately chemolithoautotrophic, moderately halophilic, thiodenitrifying Gammaproteobacteria from hypersaline habitats. Int. J. Syst. Evol. Microbiol. 2007;57:1582–1589. doi: 10.1099/ijs.0.65112-0. [DOI] [PubMed] [Google Scholar]
- 57.Polymenakou PN, Bertilsson S, Tselepides A, Stephanou EG. Bacterial community composition in different sediments from the Eastern Mediterranean Sea: a comparison of four 16S ribosomal DNA clone libraries. Microb. Ecol. 2005;50:447–462. doi: 10.1007/s00248-005-0005-6. [DOI] [PubMed] [Google Scholar]
- 58.Kanokratana P, Chanapan S, Pootanakit K, Eurwilaichitr L. Diversity and abundance of Bacteria and Archaea in the Bor Khlueng Hot Spring in Thailand. J. Basic Microbiol. 2004;44:430–444. doi: 10.1002/jobm.200410388. [DOI] [PubMed] [Google Scholar]
- 59.Mergeay M, Nies D, Schlegel HG, Gerits J, Charles P, Van Gijsegem F. Alcaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 1985;162:328–334. doi: 10.1128/jb.162.1.328-334.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goris J, De Vos P, Coenye T, Hoste B, Janssens D, Brim H, Diels L, Mergeay M, Kersters K, Vandamme P. Classification of metal-resistant bacteria from industrial biotopes as Ralstonia campinensis sp. nov., Ralstonia metallidurans sp. nov. and Ralstonia basilensis Steinle et al. 1998 emend. Int. J. Syst. Evol. Microbiol. 2001;51:1773–1782. doi: 10.1099/00207713-51-5-1773. [DOI] [PubMed] [Google Scholar]
- 61.Mudder TI, Whitlock JL. Strain of Pseudomonas paucimobilis. United States: 1984. [Google Scholar]
- 62.Kelly DP, Wood AP. Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2000;50:511–516. doi: 10.1099/00207713-50-2-511. [DOI] [PubMed] [Google Scholar]
- 63.Allpress JD, Mountain G, Gowland PC. Production, purification and characterization of an extracellular keratinase from Lysobacter NCIMB 9497. Lett. Appl. Microbiol. 2002;34:337–342. doi: 10.1046/j.1472-765x.2002.01093.x. [DOI] [PubMed] [Google Scholar]
- 64.Chohnan S, Nonaka J, Teramoto K, Taniguchi K, Kameda Y, Tamura H, Kurusu Y, Norioka S, Masaki T, Sakiyama F. Lysobacter strain with high lysyl endopeptidase production. FEMS Microbiol. Lett. 2002;213:13–20. doi: 10.1111/j.1574-6968.2002.tb11279.x. [DOI] [PubMed] [Google Scholar]
- 65.Hashizume H, Hirosawa S, Sawa R, Muraoka Y, Ikeda D, Naganawa H, Igarashi M. Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. J. Antibiot. 2004;57:52–58. doi: 10.7164/antibiotics.57.52. [DOI] [PubMed] [Google Scholar]
- 66.Asami H, Aida M, Watanabe K. Accelerated sulfur cycle in coastal marine sediment beneath areas of intensive shellfish aquaculture. Appl. Environ. Microbiol. 2005;71:2925–2933. doi: 10.1128/AEM.71.6.2925-2933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nunoura T, Takai K. Comparison of microbial communities associated with phase-separation-induced hydrothermal fluids at the Yonaguni Knoll IV hydrothermal field, the Southern Okinawa Trough. FEMS Microbiol. Ecol. 2009;67:351–370. doi: 10.1111/j.1574-6941.2008.00636.x. [DOI] [PubMed] [Google Scholar]
- 68.Takai K, Miyazaki M, Nunoura T, Hirayama H, Oida H, Furushima Y, Yamamoto H, Horikoshi K. Sulfurivirga caldicuralii gen. nov., sp. nov., a novel microaerobic, thermophilic, thiosulfate-oxidizing chemolithoautotroph, isolated from a shallow marine hydrothermal system occurring in a coral reef, Japan. Int. J. Syst. Evol. Microbiol. 2006;56:1921–1929. doi: 10.1099/ijs.0.64297-0. [DOI] [PubMed] [Google Scholar]
- 69.Sorokin DY, Lysenko AM, Mityushina LL, Tourova TP, Jones BE, Rainey FA, Robertson LA, Kuenen GJ. Thioalkalimicrobium aerophilum gen. nov., sp. nov. and Thioalkalimicrobium sibericum sp. nov., and Thioalkalivibrio versutus gen. nov., sp. nov., Thioalkalivibrio nitratis sp.nov., novel and Thioalkalivibrio denitrificancs sp. nov., novel obligately alkaliphilic and obligately chemolithoautotrophic sulfur-oxidizing bacteria from soda lakes. Int. J. Syst. Evol. Microbiol. 2001;51:565–580. doi: 10.1099/00207713-51-2-565. [DOI] [PubMed] [Google Scholar]
- 70.Mueller–Spitz SR, Goetz GW, McLellan SL. Temporal and spatial variability in nearshore bacterioplankton communities of Lake Michiga. FEMS Microbiol. Ecol. 2009;67:511–522. doi: 10.1111/j.1574-6941.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 71.Weon HY, Kim BY, Hong SB, Jeon YA, Kwon SW, Go SJ, Koo BS. Rhodanobacter ginsengisoli sp. nov. and Rhodanobacter terrae sp. nov., isolated from soil cultivated with Korean ginseng. Int. J. Syst. Evol. Microbiol. 2007;57:2810–2813. doi: 10.1099/ijs.0.65018-0. [DOI] [PubMed] [Google Scholar]
- 72.Abulencia CB, Wyborski DL, Garcia JA, Podar M, Chen W, Chang SH, Chang HW, Watson D, Brodie EL, Hazen TC, Keller M. Environmental whole-genome amplification to access microbial populations in contaminated sediments. Appl. Environ. Microbiol. 2006;72:3291–3301. doi: 10.1128/AEM.72.5.3291-3301.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.da CJE, Marsh TL, Tiedje JM, de SMFM. Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J. 2009;3:1004–1011. doi: 10.1038/ismej.2009.47. [DOI] [PubMed] [Google Scholar]
- 74.Price MN, Dehal PS, Arkin AP. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 2008;9:R4. doi: 10.1186/gb-2008-9-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]