

Abstract
Three patients belonging to two families presented with a psychomotor-dysmorphism syndrome including postnatal growth deficiency and major spondylo-, epi-, and metaphyseal skeletal involvement. Other features were muscular hypotrophy, fat excess, partial growth hormone deficiency, and, in two of the three patients, episodes of unexplained fever. Additional investigations showed mild to moderate increases of serum transaminases (particularly of aspartate transaminase (AST)), creatine kinase (CK), and lactate dehydrogenase (LDH), as well as decreased coagulation factors VIII, IX, XI, and protein C. Diagnostic work-up revealed a type 2 serum transferrin isoelectrofocusing (IEF) pattern and a cathodal shift on apolipoprotein C-III IEF pointing to a combined N- and O-glycosylation defect. Known glycosylation disorders with similar N-glycan structures lacking galactose and sialic acid were excluded. Through a combination of homozygosity mapping and expression profiling, a deep intronic homozygous mutation (c.792 + 182G>A) was found in TMEM165 (TPARL) in the three patients. TMEM165 is a gene of unknown function, possibly involved in Golgi proton/calcium transport. Here we present a detailed clinical description of the three patients with this mutation. The TMEM165 deficiency represents a novel type of CDG (TMEM165-CDG). This disorder enlarges the group of CDG caused by deficiencies in proteins that are not specifically involved in glycosylation but that have functions in the organization and homeostasis of the intracellular compartments and the secretory pathway, like COG-CDG and ATP6V0A2-CDG.
Introduction
Congenital disorders of glycosylation (CDG) are genetic defects of glycan synthesis and glycan attachment to proteins and lipids. Their number is steadily growing with some 50 CDG having been reported (Jaeken 2010, 2011; Rafiq et al. 2011; Saigoh et al. 2011; Baasanjar et al. 2011; Hennet 2012). Most of them are multisystem diseases, but skeletal abnormalities are an important/predominant clinical feature in only about 20 % of the known CDG. We present three patients, including a brother and sister, with a novel hypoglycosylation disorder characterized by a psychomotor retardation-dysmorphism syndrome with postnatal growth deficiency and major spondylo-, epi-, and metaphyseal skeletal involvement. This paper describes the clinical and laboratory findings in detail. The biochemical and molecular data are reported elsewhere (Foulquier et al. 2012).
Patients and Methods
Patients
Patient 1
This boy is the first child of unrelated Ashkenazi parents originating from Georgia. The second child is a healthy girl. The third child, a girl, was similarly affected and will be described in this paper as patient 2. Patient 1 was born at the postmenstrual age of 40 weeks after a normal pregnancy. Birth weight was 3,250 g (50th centile), length 50 cm (25th centile), and head circumference 36 cm (50th centile). He presented at the age of 5 weeks with feeding problems and failure to thrive due to cow’s milk protein allergy. Growth delay was noted within months. At 19 months of age, length was 72 cm (3rd centile is 73 cm), weight 10.7 kg (10th centile), and head circumference 48.5 cm (50th centile). Fat excess, joint hyperlaxity, hypotonia, muscle weakness, and hepatosplenomegaly were noted during the first years. Psychomotor milestones were delayed: unsupported sitting at 9 months, independent walking at 2 years, and first words at 18 months. He was first seen in our hospital at the age of 11 years because of generalized epi-metaphyseal dysplasia and joint destruction tentatively diagnosed as Desbuquois syndrome. He presented with short stature (but normal head growth), generalized adiposity, and muscular hypotrophy. He had a waddling gait and was dysmorphic (midface hypoplasia, internal strabism of the right eye, moderately high arched palate, small teeth, low-set and stiff auricles, pectus carinatum, dorsolumbar kyphosis and severe sinistroconvex scoliosis, short distal phalanges, genua vara, pedes planovalgi (Fig. 1)). Additional clinical features included rapidly growing dense hair, long and dense eyelashes, delayed dentition, a hoarse voice, joint hyperlaxity (particularly of the interphalangeal joints) except for mild extension deficit of elbows and knees and hip abduction of 45°, joint “cracking” when walking, and generalized hyporeflexia and hypotonia.
Fig. 1.
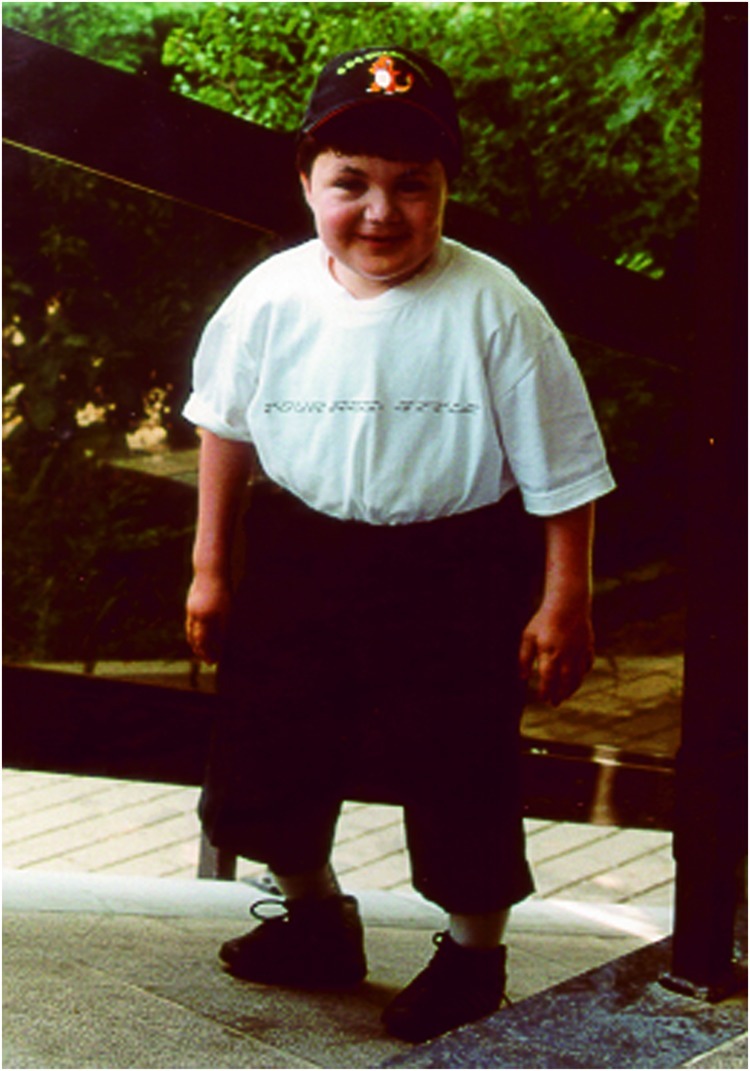
Photograph of patient 1
Laboratory investigations showed normal levels of serum alkaline phosphatases, calcium, phosphate, osteocalcine, 1,25-hydroxyvitamin D, immunoglobulin G, blood clotting factors II, V, X, XII, von Willebrand factor antigen, von Willebrand factor Ristocetine cofactor, and antithrombin, and increases of serum AST, ALT, CK, and LDH. There was a mild to moderate decrease of cholinesterase, and of clotting factors VIII, IX, XI, protein C, and protein S (Table 1). At age 16 years, there was an onset of adrenarche (circulating de-hydro-epi-androsterone-sulfate 52.4 μg/dL, normal for age being 180–350 μg/dL) but not yet of puberty (circulating luteinizing hormone 0.4 IU/L (normal 1.7–8.6 IU/L), follicle stimulating hormone 0.3 IU/L (nl 1.2–7.7), total testosterone 25 ng/dL (nl 300–1,000), free testosterone 0.27 ng/dL (nl 5–20), sex hormone binding globuline 2.58 μg/dL (nl 0.7–1.6)).
Table 1.
Abnormal results of serum parameters in the three patients
| Patient 1 | Patient 2 | Patient 3 | Normal | |
|---|---|---|---|---|
| AST (U/L) | 195–357 | 229–528 | 224–459 | <32 |
| ALT (U/L) | 43–54 | 32–59 | 32–104 | <31 |
| CK (U/L) | 1,376–1,915 | 297–2,546 | 319–918 | <145 |
| LDH (U/L) | 540–703 | 749–608 | 587–967 | 240–480 |
| Factor VIII (%) | 45 | 44 | 29 | 50–150 |
| Factor IX (%) | 63 | 42 | 30 | 70–130 |
| Factor XI (%) | 45 | 44 | 48 | 70–130 |
| Protein C (%) | 66 | 48 | 67 | 70–150 |
Technical investigations: Imaging of the skeleton showed osteoporosis and important epi- and metaphyseal dysplasia with broad metaphyses, irregular epiphyses, and thin bone cortex. The vertebrae were flattened and beaked and the phalanges were broad (Fig. 2). On echocardiography heart morphology and function were normal. Ophthalmological examination revealed a right convergent strabism and “in fundo” temporal epithelial pigment alterations, which were more pronounced in the left eye. MRI of the brain showed some atrophy, enlarged ventricles, periventricular and subcortical white matter abnormalities, as well as relative hypoplasia of the anterior pituitary, and absence of the normal hyperintensity of the posterior pituitary. A skin biopsy was normal on histology as well as on electronmicroscopy.
Fig. 2.

Radiological presentation of the skeleton of patient 1: (a) full spine at 20 years; (b) knee at 14 years 4 months; (c) elbow at 15 years 6 months; (d) hands at 15 years 6 months
Tentative treatment with growth hormone (28 μg/kg/day) in late prepuberty did normalize the circulating levels of IGF-I (from <75 μg/L to >200 μg/L) and did reduce total-body fat fraction from 28.5 % to 22.3 % within 1 year, but failed to elicit a detectable increment of growth velocity (possibly due to a concomitant aggravation of scoliosis) and was thus halted after 2 years. At age 21, the young man has a short stature (122.5 cm, Z-score −10) but a normal head circumference (55 cm, between 10th and 25th centiles) (Fig. 3).
Fig. 3.

Growth curves showing the evolution of length, weight, and head circumference in patients 1 and 3 (dotted lines); p3 is 3rd centile (full line), p50 is 50th centile (full line)
Patient 2
This girl was the third child of unrelated Ashkenazi parents from Georgia and the sibling of patient 1. Pregnancy and delivery were uneventful. Hypotonia and hip dysplasia were noted in early infancy.
She was admitted at age 5 months because of psychomotor delay, hypotonia, vomiting, and an infection-related phase of respiratory insufficiency. The girl had ptosis of the right upper eyelid, torticollis toward the left, external strabism of the left eye, and had rather large lowly implanted ears, a long filtrum, a high arched palate, a short and broad neck, a broad thorax with increased nipple distance, a sacral dimple, absent second-toe nails, hypotonia, hyporeflexia, joint hyperlaxity, right hip dysplasia, and hepatosplenomegaly (respectively, 2 and 3 cm below the costal margin).
At 11 months she was readmitted because of recurrent, unexplained fever up to 40°C during 4 months. She was a rather obese baby with weight 8.81 kg (between 25th and 50th centiles), length 68 cm (between 3rd and 10th centiles), and head circumference 44.6 cm (25th centile). She could sit unsupported for a few moments and roll over from the side to the prone or supine position, but she was not babbling. She had a large fontanel. The abdomen was slightly enlarged with liver and spleen edge at 2 cm below the costal margin. No cause for the recurrent fever was identified.
At 14 months she was admitted with a pyelonephritis. Her weight was then 8.5 kg (10th centile), and her length and head circumference were virtually the same as at 11 months (length Z-score −3.6, head circumference on 10th centile). One month later she died in an enterocolitis-related episode of shock.
Laboratory investigations showed normal results for the peripheral blood count, serum cholesterol (total, HDL-, and LDL-), lactate, albumin, amino acids, thyroxine, thyroid stimulating hormone, thyroxine-binding globuline, alkaline phosphatase, osteocalcine, arylsulfatase A, PT, APTT, clotting factors V, VII, X, XII, von Willebrand factor antigen and von Willebrand factor Ristocetine cofactor, protein S, platelet aggregation and platelet adhesion, as well as the immunoglobulins A, G, M, and D. Urinary amino acids, organic acids, and oligosaccharides were normal. Abnormal results included increases of serum AST, ALT, CK, LDH, and cerebrospinal fluid protein (566 mg/L; normal 120–450). There was a slight decrease of serum cholinesterase (6.3 kU/L; normal range 7–19) and of blood clotting factors VIII, IX, XI, antithrombin, and protein C (Table 1).
Technical investigations: Abdominal ultrasound examination was normal except for splenomegaly. Echocardiography was normal. Radiological examination of the skeleton showed generalized osteoporosis, discrete irregular metaphyses of the long bones, discrete plathyspondyly, broad iliac wings, horizontal acetabular roofs, and subluxation of the right femur. Ophthalmological investigation showed macular epithelial pigment alterations. Electrophysiological examination revealed normal sensory nerve conduction, whilst motor nerve conduction showed normal velocities but decreased amplitudes. On electromyography there were no active signs of denervation but sequelae suggestive of past denervation. Muscular biopsy showed normal histological and ultrastructural findings.
Patient 3
This boy was the fourth child of unrelated Ashkenazi parents from Georgia. This family was not related to the previously mentioned family. He was born after a normal 40 weeks pregnancy with birth weight of 3,360 g (25th centile) and length of 49 cm (between 3rd and 10th centiles). Hepatomegaly, a small open ductus Botalli and a small patent foramen ovale, were noted shortly after birth. The neonatal phase was complicated by blepharitis of the right eye, feeding difficulties (attributed to cow’s milk intolerance), diarrhea, and irritability with opisthotonus. The boy presented craniofacial dysmorphism with relative macrocephaly, tongue protrusion, anti-mongoloid slanting of the eyes, a flat nose, and relatively large and posteriorly rotated ears. At age 3 months, he was admitted because of unexplained respiratory problems (restrictive lung pathology) for which long-term invasive ventilation via tracheostomy was started. Hypotonia, muscular weakness, body adiposity, joint hyperlaxity (with cracking noise on mobilization), and brisk tendon reflexes were noted. He had regularly bouts of fever up to 38.5 °C without evidence for infection. At 14 months he developed epilepsy, responding to levetiracetam. This could be be stopped at age 2 years 10/12. The mechanical ventilation could be stopped at age 3 and the tracheostomy was closed at age 4. At age 6 years, he does not follow normally with his eyes, does not grasp for objects, and cannot sit unsupported; body length SDS is approximately −7 but head circumference is normal (49.9 cm, 25th centile) (Fig. 3). He has a distended, tympanic abdomen with liver edge at 3 cm and spleen at 2 cm below the costal margin.
Laboratory investigations showed normal serum calcium, phosphate, alkaline phosphatases, osteocalcine, arylsulfatase A, prolactin, TSH, free T4, PT, aPTT, and cholinesterase. Abnormal results included increases of serum AST, ALT, CK, LDH, and decreases of serum haptoglobin (<0.2 g/L; normal range 0.3–2), and of clotting factors VIII, IX, XI, protein C, and protein S (Table 1).
Tentative treatment with growth hormone (30 μg/kg/day, initiated at age 18 months) did normalize the circulating levels of IGF-I (from 35 μg/L to 59 μg/L) and did reduce total-body fat fraction from 27.4 % to 18.9 % within 5 months, but failed to elicit a detectable increment of growth velocity or a detectable improvement of respiratory status, and was thus halted.
Technical investigations: radiology of the skeleton at age 1 year revealed generalized osteopenia, a J-like sella turcica, hypoplasia of the skull base, mild anterior beaking of vertebrae D11, D12, L1, and L2, broad radial and ulnar metaphyses, strongly underdeveloped carpal bones, plump and broad phalanges, horizontal acetabula, very discrete opacification of the right proximal femur epiphysis, no ossification of the left proximal femur epiphysis, broad proximal femur metaphyses, broad metaphyses and strongly underdeveloped distal femoral and proximal tibial epiphyses (Fig. 4). Echocardiography at 14 months was normal except for a small pericardial effusion. Ophthalmological examination, electromyography (26 months), and motor and sensory nerve conduction velocities were normal. Brain MRI showed evidence for de- or dysmyelination and could not visualize the neural pituitary gland.
Fig. 4.

Radiological presentation of the skeleton of patient 3: (a) knee at the age of 1 year; (b) knee at 5 years 7 months; (c) lumbar spine at 1 year; (d) lumbar spine at 4 years; (e) foot at 22 m; (f) elbow at 3 years 10 months; (g) hand and wrist at 1 year; (h) hand and wrist at 3 years 6 months. See text for description
Methods
Isoelectrofocusing (IEF) of serum transferrin was performed as described (Jaeken et al. 1984).
IEF of serum apolipoprotein C-III was performed as described (Wopereis et al. 2003).
Transferrin N-glycan analysis by MALDI TOF MS was performed as described in Sturiale et al. 2008 and Papac et al. 1996 (data not shown).
Results
IEF of serum transferrin shows a type 2 pattern: decrease of 6-, 5-, and 4-sialotransferrin and increase of 3-, 2-, 1-, and 0-sialotransferrin (Fig. 5)
IEF of apolipoprotein C-III shows a decrease of the monosialo-apo C-III and an increase of the asialo-apo C-III (Fig. 5)
Serum transferrin glycan analysis shows an increase of the biantennary glycan lacking sialic acid, and an abnormal biantennary glycan lacking sialic acid and galactose. In addition there is an increased fucosylation (data not shown)
Fig. 5.

Serum transferrin IEF and ApoC-III IEF in the three patients compared to a control. Sialo fractions (0 to 6 and 0 to 2, respectively) are indicated on the left. C is control; P1 is patient 1; P2 is patient 2, P3 is patient 3
Discussion
The described patients show a congenital disorder of glycosylation with a type 2 serum transferrin pattern and a cathodal shift of serum apoC-III pointing to a combined N- and O-protein glycosylation defect. The known glycosylation disorders with a type 2 serum transferrin IEF and similar N-glycan structures (such as the COG-CDG) were excluded by mutation analysis or linkage analysis.
Common clinical features include moderate psychomotor retardation, feeding problems (in infancy), facial dysmorphism with low-set ears, hypotonia, joint hyperlaxity, hepatosplenomegaly, osteoporosis, and spondylo-, epi- and metaphyseal dysplasia aggravating with age. After infancy (patients 1 and 3), midface hypoplasia, muscular hypotrophy, fat excess, growth retardation, and partial growth hormone deficiency completed this picture. An unusual symptom in patients 2 and 3 (seen also in some COG-CDG patients; Zeevaert et al. 2008) was recurrent fever without evidence for infection. Features only present in patient 3 (family 2) were respiratory problems due to unexplained restrictive lung pathology and transient epilepsy. It remains uncertain whether these features are secondary to the CDG.
The main biochemical findings in all three patients were an increase of serum AST (moderate), ALT (very mild), CK (moderate), and LDH (mild), as well as a decrease of coagulation factors VIII, IX, XI, and protein C. Ophthalmological examination in patients 1 and 2 showed macular epithelial pigment alterations, and was normal in patient 3. Common findings on brain MRI in patients 1 and 3 were white matter abnormalities and absent visualization of the neural pituitary gland.
Skeletal manifestations are an important/predominant part of the phenotype in only about 20 % of the known CDG: EXT1/EXT2-CDG (Jennes et al. 2009), B4GALT7 (Seidler et al. 2006), B3GAT3-CDG (Baasanjar et al. 2011), GALNT3-CDG (Chefetz and Sprecher 2009), SLC35D1-CDG (Hiraoka et al. 2007), LFNG-CDG (Sparrow et al. 2006), PIGV-CDG (Horn et al. 2011), autosomal recessive cerebrocostomandibular like syndrome due to COG1-CDG (Zeevaert et al. 2009), and ATP6V0A2-CDG (Guillard et al. 2009). The spectrum of these skeletal manifestations is very broad which can be explained by the fact that glycosylation is an important posttranslational modification of numerous proteins, including extracellular matrix proteins and proteins involved in bone metabolism, for example, collagen I (Coman et al. 2007) and FGFR-3 (Winterpacht et al. 2000). The function of these proteins can be affected by a changed or absent glycosylation site. For example, a mutation affecting a putative glycosylation site of FGFR-3 leads to hypochondroplasia. Other hypotheses about the pathophysiology of bone abnormalities in CDG include lysosomal dysfunction (Garel et al. 1998) and polymorphisms in alternative pathways (Coman et al. 2008).
Thus, the bone anomalies in the three patients are unprecedented among CDG patients. Clinically, their skeletal phenotype can be classified in group 13 (among 40 groups) of the classification of genetic skeletal disorders (revision 2010), namely, the spondylo-epi-(meta)-physeal dysplasias (Warman et al. 2011).
Eventually, the molecular defect in these patients was identified through a combination of homozygosity mapping and expression profiling. The three patients are homozygous for a deep intronic splice mutation in TMEM165 (TPARL) c.792 + 182G>A (Foulquier et al. 2012). This mutation was not identified in 100 Jewish control alleles.
This disorder enlarges the group of CDG that results from deficiencies in proteins that are not specifically involved in glycosylation but that have functions in the organization and homeostasis of the intracellular compartments and the secretory pathway. Other examples of these are the COG-CDG (Foulquier 2009), ATP6V0A2-CDG (Guillard et al. 2009), and SEC23B-CDG (Bianchi et al. 2009; Schwarz et al. 2009). We propose to call this CDG subgroup “CDG plus”.
Abbreviations
- ALT
Alanine transaminase
- AST
Aspartate transaminase
- CDG
Congenital disorder(s) of glycosylation
- CK
Creatine kinase
- IEF
Isoelectrofocusing
- LDH
Lactate dehydrogenase
Take Home Message
Bone dysplasia is the main clinical feature of three patients with a novel congenital disorder of glycosylation due to a mutation in TMEM165.
Footnotes
Competing interests: None declared
References
- Baasanjar S, Al-Gazali L, Hashiguchi T, et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89:15–27. doi: 10.1016/j.ajhg.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi P, Fermo E, Vercellati C, et al. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 2009;30:12932–1298. doi: 10.1002/humu.21077. [DOI] [PubMed] [Google Scholar]
- Chefetz I, Sprecher E. Familial tumoral calcinosis and the role of O-glycosylation in the maintenance of phosphate homeostasis. Biochim Biophys Acta. 2009;1792:847–852. doi: 10.1016/j.bbadis.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coman D, Bostock D, Hunter M, et al. Primary skeletal dysplasia as a major manifesting feature in an infant with congenital disorder of glycosylation type Ia. Am J Med Genet A. 2007;146A:389–392. doi: 10.1002/ajmg.a.32119. [DOI] [PubMed] [Google Scholar]
- Coman D, Irving M, Kannu P, Jaeken J, Savarirayan R. The skeletal manifestations of the congenital disorders of glycosylation. Clin Genet. 2008;73:507–515. doi: 10.1111/j.1399-0004.2008.01015.x. [DOI] [PubMed] [Google Scholar]
- Foulquier F. COG defects, birth and rise! Biochim Biophys Acta. 2009;1792:896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- Foulquier F, Amyere M, Jaeken J et al (2012) TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet 91:15–26 doi:10.1016/j.ajhg.2012.05.002 [DOI] [PMC free article] [PubMed]
- Garel C, Baumann C, Besnard M, et al. Carbohydate-deficient glycoprotein syndrome type I: a new cause of dysostosis multiplex. Skeletal Radiol. 1998;27:43–45. doi: 10.1007/s002560050335. [DOI] [PubMed] [Google Scholar]
- Guillard M, Dimopoulou A, Fischer B, et al. Vacuolar H+-ATPase meets glycosylation in patients with cutis laxa. Biochim Biophys Acta. 2009;1792:903–914. doi: 10.1016/j.bbadis.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Hennet T (2012) Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim Biophys Acta 1820:1306–1317 [DOI] [PubMed]
- Hiraoka S, Furuichi T, Nishimura G, et al. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat Med. 2007;13:1363–1367. doi: 10.1038/nm1655. [DOI] [PubMed] [Google Scholar]
- Horn D, Krawitz P, Mannhardt A, Korenke GC, Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am J Med Genet A. 2011;155:1917–1922. doi: 10.1002/ajmg.a.34102. [DOI] [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis. 2011;34:853–858. doi: 10.1007/s10545-011-9299-3. [DOI] [PubMed] [Google Scholar]
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized syndrome. Clin Chim Acta. 1984;144:245–247. doi: 10.1016/0009-8981(84)90059-7. [DOI] [PubMed] [Google Scholar]
- Jennes I, Pedrini E, Zuntini M, et al. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb) Hum Mut. 2009;30:1620–1627. doi: 10.1002/humu.21123. [DOI] [PubMed] [Google Scholar]
- Papac DI, Wong A, Jones AJ. Analysis of acidic oligosaccharides and glycopeptides by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Chem. 1996;68:3215–3223. doi: 10.1021/ac960324z. [DOI] [PubMed] [Google Scholar]
- Rafiq MA, Kuss AW, Puettmann L, et al. Mutations in the alpha 1,2-mannosidase gene, MAN1B1, cause autosomal recessive intellectual disability. Am J Hum Genet. 2011;89:176–182. doi: 10.1016/j.ajhg.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigoh K, Izumikawa T, Koike T, Shimizu J, Kitagawa H, Kusunoki S. Chondroitin beta-1,4-N-acetylgalactosaminyltransferase-1 missense mutations are associated with neuropathies. J Hum Genet. 2011;56:143–146. doi: 10.1038/jhg.2010.148. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Iolasco A, Verissimo F, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- Seidler DG, Faijaz-Ul-Haque M, Hansen U, et al. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (beta4GalT-7) J Mol Med. 2006;84:583–594. doi: 10.1007/s00109-006-0046-4. [DOI] [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Wouters MA, et al. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 2006;78:28–37. doi: 10.1086/498879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturiale L, Barone R, Palmigiano A, Ndosimao CN, Briones P, Adamowicz M, Jaeken J, Garozzo D. Multiplexed glycoproteomic analysis of glycosylation disorders by sequential yolk immunoglobulins immunoseparation and MALDI-TOF MS. Proteomics. 2008;18:3822–3832. doi: 10.1002/pmic.200700496. [DOI] [PubMed] [Google Scholar]
- Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterpracht A, Hilbert K, Stelzer C, et al. A novel mutation in FGFR-3 disrupts a putative N-glycosylation site and results in hypochondroplasia. Physiol Genomics. 2000;2:9–12. doi: 10.1152/physiolgenomics.2000.2.1.9. [DOI] [PubMed] [Google Scholar]
- Wopereis S, Grünewald S, Morava E, et al. Apolipoprotein C-III isofocusing in the diagnosis of genetic defects in O-glycan biosynthesis. Clin Chem. 2003;49:1839–1845. doi: 10.1373/clinchem.2003.022541. [DOI] [PubMed] [Google Scholar]
- Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the conserved oligomeric Golgi (COG) complex define a novel group of congenital disorders of glycosylation. Mol Genet Metab. 2008;93:15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]
- Zeevaert R, Foulquier F, Dimitrov B, et al. Cerebrocostomandibular-like syndrome and a mutation in the conserved oligomeric Golgi complex, subunit 1. Hum Mol Genet. 2009;18:517–524. doi: 10.1093/hmg/ddn379. [DOI] [PubMed] [Google Scholar]