

Abstract
In 2010, we reported the successful clinical outcome related to a 20-month course of intravenous, cyclical ceftriaxone, in a patient with adult-onset Alexander’s disease. We now provide evidence that the progression of the patient’s signs/symptoms was halted and reversed with a 4-year-long extension of the trial.
The patient’s clinical signs/symptoms were evaluated before the start and every 6 months for 6 years. For the early 2 years, without therapy, and for the following 4 years, after intravenous ceftriaxone 2 g daily, for 3 weeks monthly during the initial 4 months, then for 15 days monthly.
Gait ataxia and dysarthria were assessed clinically on a 0 to 4 scale. Palatal myoclonus and nystagmus/oscillopsia were monitored by videotape and a self-evaluation scale. The degree of disability, measured by a modified Rankin scale, and the brain MRI were periodically evaluated.
Before ceftriaxone therapy, in a 2-year period, gait ataxia and dysarthria worsened from mild to marked, palatal myoclonus spread from the soft palate to lower facial muscles, and the patient complained of oscillopsia. After 4 years of ceftriaxone therapy, gait ataxia and dysarthria improved, from marked to mild at clinical rating scales. The palatal myoclonus was undetectable; the patient did not complained of oscillopsia and declared a progressively better quality of life. Ceftriaxone was safe.
This case report provides Class IV evidence that intravenous cycles of ceftriaxone may halt and/or reverse the progression of neurodegeneration in patients with adult-onset Alexander’s disease and may significantly improve their quality of life.
Introduction
Alexander’s disease (AxD) is a rare, usually fatal, primary disorder of astroglial cells in the central nervous system (CNS) related to dominant mutations in the gene encoding the type III intermediate filament protein, glial fibrillary acidic protein (GFAP) (Brenner et al. 2001; Mignot et al. 2004; Li et al. 2005). The prevalent pathophysiological hypothesis regarding AxD is based on the occurrence of a toxic gain-of-function of mutated GFAP, which causes intracytoplasmic aggregates in astrocytes (Rosenthal fibers), containing GFAP, αB-Crystallin, the heat shock protein 27, and ubiquitin (Mignot et al. 2004; Li et al. 2005), and on the occurrence of excitotoxicity related to impairment of the buffering capacity of dystrophic astrocytes and of their ability to metabolize extracellular glutamate (Mignot et al. 2004; Tian et al. 2010). Therefore, in AxD, lowering the production of GFAP and promoting its degradation and/or the enhancement of the astroglial glutamate uptake from the extracellular fluid may have potential therapeutic effects. No treatment is at present available for this neurodegenerative disorder, although recent data, in vitro and in animal models of AxD, indicate that drugs such as the β-lactam antibiotic ceftriaxone and the tricyclic antidepressant clomipramine may have potential therapeutic effects (Bachetti et al. 2010; Cho et al. 2010). In particular, clomipramine lowers the production of GFAP in an animal model of AxD and delays the appearance of aggregates in astrocytes from the animal model (Cho et al. 2010). Ceftriaxone enhances the degradation of GFAP aggregates in a cellular model of AxD (Bachetti et al. 2010), and may also counteract neurodegeneration due to glutamatergic excitotoxicity, based on its ability to increase glutamate transporter subtype 1 (GLT-1) activity in astrocytes (Rothstein et al. 2005). Previously, we reported the successful clinical outcome related to a 20-month course of intravenous, cyclical ceftriaxone in a patient with an adult form of AxD and a rapidly progressive clinical course (Sechi et al. 2010a). Here we studied the tolerability and therapeutic effects of cycles of ceftriaxone in this patient at a 4-year follow-up.
Methods
The patient is a 44-year-old woman carrying the p.R70Q and p.D157N GFAP mutations with adult-onset AxD (Sechi et al. 2010a). The prominent clinical signs/symptoms were dysarthria, gait ataxia, and palatal myoclonus from the age of 38 years and evoked nystagmus/oscillopsia from the age of 40 years. We previously reported the effect of a 20-month long trial of ceftriaxone therapy. At that time, the patient declared an improvement of her clinical status, which was in agreement with clinical (ataxia, dysarthria) and electrophysiological (nystagmography recording) data (Sechi et al. 2010a). We presently report an extension of the trial encompassing a 6-year-long observation.
Gait ataxia, dysarthria, palatal myoclonus, and evoked nystagmus/oscillopsia were evaluated every 6 months over a 6-year period. For the first 2 years, without therapy, and for the following 4 years, after intravenous ceftriaxone 2 g daily, for 3 weeks monthly during the initial 4 months, then for 15 days monthly. Among the ceftriaxone cycles, a probiotic preparation was administered by mouth once daily: FlorVis-gg, containing 6 milliards of Lactobacillus rhamnosus. The patient’s body weight was 60 kg. Her Mini-Mental State Examination score was 30. Intravenous ceftriaxone was administered by a chronic subcutaneously implanted port on the chest.
Gait ataxia and dysarthria were assessed clinically on a 0 to 4 scale (0 = no disturbance, 1 = mild, 2 = moderate, 3 = marked, 4 = severe). In particular, speech was assessed during normal conversation for at least 10 min, and the severity of dysarthria evaluated according to the following rating scale:
0: no disturbance.
1: mild. The speech is impaired, but all the words are understandable.
2: moderate. Occasional words difficult to understand.
3: marked. Many words difficult to understand.
4: severe. Only single words are understandable.
For evaluating gait ataxia, instead, the patient was asked to walk at a safe distance parallel to a wall, including a turn around to face the opposite direction of gait (half-turn), and to walk in tandem without support. The severity of gait ataxia was rated as follows:
0: no disturbance.
1: mild. Slight difficulties, only evident when walking in tandem.
2: moderate. Definite staggering, difficulties in half-turn but without support.
3: marked. Marked staggering, intermittent support of the wall required.
4: severe. Walking is only possible with strong support, stroller, or accompanying person.
Palatal myoclonus was monitored by videotape and the progressive involvement of contiguous muscles noticed. Videotape examinations were conducted in the same physical environment and conditions and with the same video equipment. Oscillopsia was monitored by a self-evaluation scale (severe, moderate, mild, no oscillopsia). The degree of disability was measured, every year, for 6 years, by a modified Rankin scale (van Swieten et al. 1988). The scale runs from 0–6, running from perfect health without symptoms to death. Brain MRI was evaluated about every 2 years. Periodic controls of hematological parameters, electrocardiography (ECG) and electroencephalography (EEG) were performed. The patient gave written informed consent. The study was approved by the local ethics committee.
Results
Before ceftriaxone therapy, during the first 2 years of observation, the patient’s gait ataxia and the dysarthria worsened progressively, from mild to marked at clinical rating scales, and the palatal myoclonus spread, after about 10 months, from the soft palate to lingual and lower facial muscles. At the beginning of the study, eye movement recording (EMR) was normal and the patient did not complain of oscillopsia. After about 16 months, at EMR, bilateral hypometric horizontal saccades and horizontal left gaze evoked nystagmus with waveform-type pendular-like mixed were present, and the patient complained of severe oscillopsia. After 2 years without therapy, the gait ataxia, the oscillopsia, and the dysarthria prevented a completely independent way of life (Rankin score, 3). Brain MRI revealed bilateral, high signal-intensity areas posteriorly in the periventricular white matter, pyramidal tracts of the medulla, and cerebellar white matter (Sechi et al. 2010a).
After about 2 years of ceftriaxone therapy, the severity of palatal myoclonus did not worsen; the gait ataxia and the dysarthria improved from marked to moderate at clinical rating scales. The evoked nystagmus showed a reduced amplitude in comparison with the previous recordings, with improvement in the ability to read or watch television. The patient rated her oscillopsia as mild. The patient rated herself as improved and declared a better quality of life (Rankin score, 1–2) (Sechi et al. 2010a).
After about 4 years of ceftriaxone therapy, the gait ataxia and the dysarthria showed a definite improvement from marked to mild at clinical rating scales. The palatal myoclonus, after about 3 years, disappeared from lower facial muscles and tongue, and after about 4 years it was undetectable (videos 1, 2). The patient did not complain of oscillopsia, rated herself as definitively improved, and declared a progressively better quality of life (Rankin score, 1) (Fig. 1). Importantly, different examiners blinded to the treatment, at the different time points, agreed for a progressive improvement of the gait ataxia, dysarthria, and palatal myoclonus in the patient. On follow-up examination, the signal-intensity changes on brain MRI did not reverse but did not progress further or show atrophy in involved areas (Fig. 2a–d). Ceftriaxone did not have obvious, major toxicity in the patient. After 7 months of therapy, the patient had a mycotic vaginal infection treated with miconazole. After 3.5 years, mainly during ceftriaxone cycles, the patient occasionally complained of single, right or left myoclonic jerks in upper limbs, about seven times a week. EEG showed a normal background rhythm without epileptiform activity. The patient refused any therapy for myoclonic jerks. Periodic controls, of white and red blood cell counts, glucose, creatinine, serum electrolytes, liver function tests, ECG, and EEG were normal.
Fig. 1.
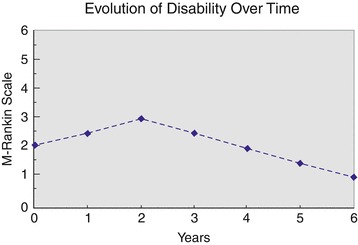
Evolution of the degree of disability over time: 0–2 years, before treatment; 2–6 years, after ceftriaxone treatment
Fig. 2.
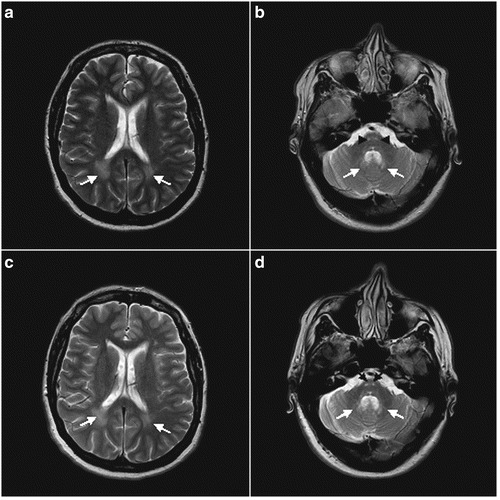
a–d: Axial T2-WMR images, at the beginning of ceftriaxone therapy (a, b) and after about 4 years of ceftriaxone therapy (c, d). Bilateral high signal-intensity areas are seen posteriorly in the periventricular white matter (arrows) (a, c); MR scan obtained at a lower level shows involvement of the descending pyramidal tracts of the medulla (arrowheads) and deep cerebellar white matter (arrows) (b, d). On follow-up examination, the signal-intensity changes did not reverse but did not progress further, or show atrophy in involved areas
Discussion
The available literature data indicate that in patients with adult-onset AxD, the clinical course is usually that of a progressive neurodegenerative disease without spontaneous remission (Schwankhaus et al. 1995; Balbi et al. 2010), as also shown by the evaluation of our patient during the first 2 years of observation without therapy. Thus, it seems very unlikely that the progressive improvement of the patient over a 4-year period, after ceftriaxone therapy, may be due to the natural course of the disease.
This case report provides Class IV evidence that parenteral cycles of ceftriaxone were able to halt and reverse the progression of the disease and to improve significantly the quality of life in a patient with adult-onset AxD and a rapidly progressive clinical course. Importantly, a definite improvement was achieved slowly for all the signs evaluated, from few months to 3 years, with a different timescale which seems related to the time necessary to the development and progression of the different signs, as exemplified from palatal myoclonus. On follow-up examination, brain MRI changes did not progress further, or show atrophy in involved areas. Because the available literature indicates that in patients with AxD the brain MRI signal intensities are not static but may vary in time and progress to atrophy in involved areas (Sawaishi 2009), this finding also may be indicative of a therapeutic effect of ceftriaxone in our patient with AxD. At the used dosages, cyclical administration of ceftriaxone was safe. Some adverse effects, as the self-limiting myoclonic jerks, indicate that ceftriaxone may lower the epileptogenic threshold. This adverse event needs long-term supervision, and an eventual lowering of ceftriaxone dosages, mainly in the infantile form of AxD, which is frequently characterized by the occurrence of epileptic seizures (Li et al. 2005).
Recent, essential advances in the understanding of the pathophysiology of AxD indicate that a dynamic and reversible aggregation of mutated GFAP may occur (Mignot et al. 2004; Mignot et al. 2007), and that in this pathology at least three main biochemical mechanisms play a fundamental role in promoting neurodegeneration: (1) The key initiating event is the expression and accumulation in astrocytes of mutant GFAP above a toxic threshold, with consequent, progressive astroglial damage (Messing et al. 1998). (2) The decrease of the cytoplasmic content of the small heat-shock proteins (sHPSs) HSP27 and αB-Crystallin in astrocytes, due to their sequestration in GFAP aggregates (Mignot et al. 2004; Der Perng et al. 2006). Indeed, it has been shown that overexpression of these sHPSs in astrocytes promotes dissolution of GFAP aggregates (Tang et al. 2010). (3) The impairment of the buffering capacity of dystrophic astrocytes and of their ability to metabolize extracellular glutamate, with chronic accumulation of extracellular glutamate and subsequent excitotoxicity, may play an important role in the pathogenesis of secondary oligodendrocyte and neuronal degeneration in AxD (Tian et al. 2010; Sechi et al. 2010b). Given these findings, it is conceivable that in AxD, for achieving an effective and long-term suppression of neurodegeneration, a chemical agent, or a combination of chemical agents that inhibit multiple biochemical injurious mechanisms in astrocytes may be needed. In such a scenario, ceftriaxone may be of particular interest. This β-lactam antibiotic is a safe and multipotent agent used for decades as antimicrobial (Nau et al. 1993). In 2005, ceftriaxone was suggested as a potential therapeutic agent in neurodegenerative diseases comprising glutamate-mediated toxicity, based on its ability to increase glutamate transporter subtype 1 (GLT-1) activity in astrocytes that are genetically impaired in their GLT-1 expression (Rothstein et al. 2005). In 2010, we showed that ceftriaxone is also able to successfully eliminate the toxic effects of misfolded GFAP in a cellular model of AxD by decreasing GFAP intracytoplasmic aggregates (Bachetti et al. 2010). Underlying mechanisms include mutant GFAP elimination, concurrent with upregulation of HSP27 and αB-Crystallin, polyubiquitination, enhancement of autophagy, and GFAP promoter downregulation (Bachetti et al. 2010). Interestingly, a recent study in an animal axotomy model showed that ceftriaxone was able to suppress, after 28 days of parenteral administration, the reactive increase of astroglial GFAP expression to control level, at 200 mg/kg, daily dose (Yamada and Jinno 2011). Another, very recent study in a mouse model of AxD documented that overexpression of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) dramatically reduced astrocytic GFAP content in all brain regions examined and restored body weights in R236H mice to near wild-type levels (Lapash Daniels et al. 2012). Since the astrocytic induction of Nrf2 is involved in the neuroprotective action of ceftriaxone (Lewerenz et al. 2009), this mechanism also may partly explain the therapeutic effect of this drug in AxD. Notably, in AxD the ceftriaxone activation of cellular mechanisms involved in the disposal of mutant GFAP acts in a synergistic way with its ability to counteract the chronic glutamatergic excitotoxicity in this pathology, with consequent rescue of the secondary oligodendrocytes and neuronal degeneration related to excitotoxicity (Sechi et al. 2010b).
Because AxD is rare and its presentation and course vary, evaluation of treatments in placebo-controlled trials is very difficult (Sechi et al. 2010a). Instead, prolonged, longitudinal single patient studies may be a useful approach to identify new therapeutic strategies in this disorder (Sechi et al. 2010a). The successful clinical outcome related to ceftriaxone documented in our patient highlights the possibility that this safe drug may be useful for other AxD patients and puts the relevant question whether ceftriaxone may also be useful in treating other diseases caused by intracellular accumulation and aggregation of misfolded proteins (Sechi et al. 2011), particularly other neurodegenerative diseases with astrocyte involvement.
Synopsis
Long-term intravenous cycles of ceftriaxone have a therapeutic role in adult-onset Alexander’s disease with a rapidly progressive clinical course.
GianPietro Sechi designed the study, analyzed and interpreted the data, provided the figures, wrote the manuscript, and obtained funding.
Isabella Ceccherini revised the manuscript for content.
Tiziana Bachetti revised the manuscript for content.
Giovanni A. Deiana analyzed and interpreted the data and revised the manuscript for content.
Elia Sechi revised the manuscript for content.
Pietro Balbi analyzed and interpreted the data and revised the manuscript for content.
GianPietro Sechi serves as guarantor for the article, accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Isabella Ceccherini, Tiziana Bachetti, Giovanni A. Deiana, Elia Sechi, and Pietro Balbi report no disclosures.
GianPietro Sechi received research support from the Regione Sardegna. He confirms independence from the sponsors; the content of the article has not been influenced by the sponsors.
The study was approved by the local ethics committee.
Footnotes
Competing interests: None declared
Electronic supplementary material: The online version of this article (doi:10.1007/8904_2012_180) contains supplementary material, which is available to authorized users.
References
- Bachetti T, Di Zanni E, Balbi P, et al. In vitro treatments with ceftriaxone promote elimination of mutant glial fibrillary acidic protein and transcription down-regulation. Exp Cell Res. 2010;316:2152–2165. doi: 10.1016/j.yexcr.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Balbi P, Salvini S, Fundarò C, et al. The clinical spectrum of late-onset Alexander disease: a systematic literature review. J Neurol. 2010;257:1955–1962. doi: 10.1007/s00415-010-5706-1. [DOI] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boesflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/87020. [DOI] [PubMed] [Google Scholar]
- Cho W, Brenner M, Peters N, Messing A. Drug screening to identify suppressors of GFAP expression. Hum Mol Genet. 2010;19:3169–3178. doi: 10.1093/hmg/ddq227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der Perng M, Su M, Wen SF, et al. The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and tha association of αB-crystallin and HSP27. Am J Hum Genet. 2006;79:197–213. doi: 10.1086/504411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapash Daniels CM, Austin EV, Rockney DE, et al. Beneficial effects of Nrf2 overexpression in a mouse model of Alexander disease. J Neurosci. 2012;32:10507–10515. doi: 10.1523/JNEUROSCI.1494-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Albrecht P, Tran Tien ML, et al. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- Li R, Salomons G, Johnson AB, Naidu S, et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005;2005(57):310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D. Alexander disease: putative mechanisms of an astrocytic encephalopathy. CMLS, Cell Mol Life Sci. 2004;61:369–385. doi: 10.1007/s00018-003-3143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Delarasse C, Escaich S, et al. Dynamics of mutated GFAP aggregates revealed by real-time imaging of an astrocyte model of Alexander disease. Exp Cell Res. 2007;313:2766–2779. doi: 10.1016/j.yexcr.2007.04.035. [DOI] [PubMed] [Google Scholar]
- Nau R, Prange HW, Muth P, et al. Passage of cefotaxime and ceftriaxone into cerebrospinal fluid of patients with uninflamed meninges. Antimicrob Agents Chemother. 1993;37:1518–1524. doi: 10.1128/AAC.37.7.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, et al. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sawaishi Y. Review of Alexander disease: beyond the classical concept of leukodystrophy. Brain Dev. 2009;31:493–498. doi: 10.1016/j.braindev.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Schwankhaus JD, Parisi JE, Gulledge WR, Chin L, Currier RD. Hereditary adult-onset Alexander’s disease with palatal myoclonus, spastic paraparesis, and cerebellar ataxia. Neurology. 1995;45:2266–2271. doi: 10.1212/WNL.45.12.2266. [DOI] [PubMed] [Google Scholar]
- Sechi GP, Matta M, Deiana GA, et al. Ceftriaxone has a therapeutic role in Alexander disease. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:416–417. doi: 10.1016/j.pnpbp.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Sechi GP, Balbi P, Bachetti T, Ceccherini I. Correspondence regarding: Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol. 2010;69:1271–1272. doi: 10.1097/NEN.0b013e3181fe9e86. [DOI] [PubMed] [Google Scholar]
- Sechi GP, Balbi P, Bachetti T, Ceccherini I. Safe drugs to fight mutant protein overload and alpha-1-antitrypsin deficiency. J Hepatol. 2011;55:949–950. doi: 10.1016/j.jhep.2011.03.033. [DOI] [PubMed] [Google Scholar]
- Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein αB-crystallin reverse the inhibition. J Biol Chem. 2010;285:10527–10537. doi: 10.1074/jbc.M109.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Wu X, Hagemann TL, Sosunov AA, Messing A, McKham GM, Goldman JE. Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol. 2010;69:335–345. doi: 10.1097/NEN.0b013e3181d3cb52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19:604–607. doi: 10.1161/01.STR.19.5.604. [DOI] [PubMed] [Google Scholar]
- Yamada J, Jinno S (2011) Alterations in neuronal survival and glial reactions after axotomy by ceftriaxone and minocycline in the mouse hypoglossal nucleus. Neurosci Lett. doi:10.1016/j.neulet.2011. 09.051 [DOI] [PubMed]