

Abstract
Rare loss-of-function mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene are known to dramatically decrease the catalytic activity of acid sphingomyelinase (ASM), resulting in an autosomal recessive lysosomal storage disorder known as Niemann-Pick disease (NPD) type A and B. In contrast to the general low frequency of those deleterious mutations, we found a relatively high frequency for the proposed type B NPD variant c.1460C>T (p.A487V) in our sample of 58 patients suffering from Major Depressive Disorder. We therefore investigated the biochemical consequences of this variant more closely. Our in vivo data derived from blood cell analyses indicated cellular ASM activity levels in the normal range. The secreted ASM activity levels in blood plasma were slightly lower, but still above those levels reported for type B NPD patients. In vitro expression studies of this ASM variant in different cell lines confirmed these results, showing cellular and secreted enzymatic activities equivalent to those of wild-type ASM and similar expression levels. Thus, we conclude that the ASM variant c.1460C>T (p.A487V) is not a rare missense mutation but an SMPD1 sequence variant that yields a protein with functional catalytic characteristics.
Introduction
An inherited deficiency of acid sphingomyelinase (EC 3.1.4.12; ASM) is the cause of autosomal recessive sphingolipidosis, known as Niemann-Pick disease (NPD) type A [MIM: 257200] or B [MIM: 607616] (Brady et al. 1966). Type A NPD is a severe infantile neurodegenerative disease that results in death within 3 years of life. It is also characterised by massive hepatosplenomegaly and psychomotor deterioration. In contrast, type B NPD does not display neurological involvement, but is a visceral form featuring hepatosplenomegaly and affecting the reticuloendothelial and respiratory system. Type B NPD patients can survive into adulthood (Dardis et al. 2005). The birth prevalence is estimated to be in the range of 0.5 per 100,000 (Pinto et al. 2004), with the exception of the Ashkenazi Jews, in whom the birth prevalence is approximately 3 per 100,000 (Schuchman and Miranda 1997). In addition to type A and type B, intermediate NPD phenotypes have been observed (Pavlu-Pereira et al. 2005; Wasserstein et al. 2006), leading to a proposed alternative classification system that includes four NPD types (Harzer et al. 2003).
Currently, up to 100 missense mutations in the sphingomyelin phosphodiesterase 1 gene (SMPD1; GenBank Accession Number NM_000543.4), which codes for ASM, are listed in the Human Gene Mutation Database. Novel SMPD1 mutations are usually identified from patients presenting clinical symptoms of NPD and reduced cellular ASM activity (L-ASM, lysosomal) in cultured skin fibroblasts. Putative disease-causing mutations are subsequently identified by sequencing. The secreted form of ASM in blood plasma (S-ASM) can be diagnostically used and helps to distinguish between NPD patients (S-ASM activity below 7% of normal activity; range 0.18–7%) and NPD carriers (below 41% of normal activity; range 8–41%) (He et al. 2003). There exist validated genotype–phenotype correlations, which are based on the residual cellular ASM activity levels. Residual L-ASM activity under 5% is supposed to be indicative of NPD type A (Desnick et al. 2010).
Patients suffering from Major Depressive Disorder (MDD) display significantly higher levels of ASM activity in their peripheral blood cells compared to controls, and the severity of depression and ASM activity levels are positively correlated (Kornhuber et al. 2005). To analyse the impact of sequence variations on ASM activity levels in the context of MDD, we conducted a re-sequencing analysis of SMPD1 in MDD patients. Among the identified missense variations was the single nucleotide polymorphism (SNP) c.1460C>T (rs141641266) that codes for the amino acid substitution p.A487V. This variant is a proposed type B NPD mutation and was first described in a patient with a less severe form of type B NPD characterised by organomegaly and mild pulmonary involvement (Simonaro et al. 2002).1 Within a recent exome sequencing project, the frequency of this variant was 0.4%. To clarify the biochemical consequences of the proposed type B NPD variant c.1460C>T/p.A487V, we used in vivo and in vitro approaches. The cloning of identified mutations into expression vectors and the overexpression of the mutant proteins in cultured cells provide a powerful method to determine the impact of a particular mutation on ASM activity (Dardis et al. 2005; Desnick et al. 2010; Jones et al. 2008). To date, however, most of the proposed ASM missense mutations have not been verified by expression studies, which are necessary to provide conclusive evidence of their disease-causing loss-of-function status.
In this study, we clearly showed using in vivo and in vitro approaches that the ASM variant c.1460C>T/p.A487V is not associated with decreased levels of enzymatic activity, but displays functional enzymatic characteristics.
Materials and Methods
Ethics Statement
The collection of blood samples was approved by the local Ethics Committees and conducted in concordance with the Declaration of Helsinki. Written informed consent was obtained from all participants.
Subjects
For the initial re-sequencing analysis, 58 patients suffering from MDD according to ICD-10 criteria and treated at the University Hospital Erlangen, Germany, were included. For further sequencing analyses, samples from 420 patients with affective disorders and 422 control samples were provided by the German Research Network on Depression and Suicidality. Diagnosis was confirmed by common psychiatric procedures. Additional control samples were collected and analysed for sequence variations and ASM activity levels at the University Hospital Erlangen, Germany.
Genotyping
Genomic DNA was isolated from whole blood using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). The sequence variant c.1460C>T was amplified by polymerase chain reaction (PCR) using the oligonucleotides 5′-CCCCAGATGTCTTCCTACCCC-3′ and 5′-TTCCCCTATCCCACCAACTCC-3′ and cycle sequenced on an Applied Biosystems 3730 DNA Analyzer using the same primers.
Overexpression of ASM Variants
A wild-type ASM transcript was amplified from RNA by PCR and cloned into the FLAG-N2 expression vector to create an in-frame C-terminal fusion of ASM with the 16 amino acid FLAG-tag (Rhein et al. 2012). The variants A487V and P325A were generated by site-directed mutagenesis using the QuickChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). ASM variants were transfected into HeLa cells by the calcium phosphate precipitation procedure (Rhein et al. 2012). MDCK cells were transfected using polyethylenimine (Ehrhardt et al. 2006). Cells were harvested 48 h after transfection and lysates were analysed.
Western Blot Analysis
Transfected cells were lysed in RIPA buffer and analysed for protein concentration using the BCA assay (Bio-Rad, Munich, Germany). Total protein (10 μg) was separated by 10% SDS gel electrophoresis and transferred to a PVDF-membrane (Millipore, Schwalbach/Ts, Germany). Blots were incubated with a primary mouse monoclonal anti-FLAG-antibody (1:1,000; Sigma-Aldrich, Munich, Germany), with a primary mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:200,000; Millipore, Schwalbach/Ts, Germany) and with a secondary goat anti-mouse IgG antibody, coupled to horseradish peroxidase (1:10,000; Dianova, Hamburg, Germany). Detection was performed using the enhanced chemiluminescence (ECL) Western blotting detection system (Amersham, Munich, Germany) and visualised on a high-sensitivity camera device (Fluor-S-Max, Bio-Rad, Munich, Germany) (Rhein et al. 2012).
In Vitro Determination of ASM Activity
L-ASM and S-ASM activity from primary samples and from cell lysates was determined using the fluorescent substrate BODIPY-C12-sphingomyelin (Invitrogen, Darmstadt, Germany). The method was validated and described in detail in Reichel et al. (2011). Briefly, 2 μl of cell lysate (typically corresponding to 0.5–2 μg of protein), 10 μl of serum-free conditioned medium or 1.5 μl blood plasma were added to 116 pmol of fluorescent substrate in sodium acetate buffer (200 mM sodium acetate pH 5.0, 500 mM NaCl and 0.2% NP-40) in a total volume of 100 μl. For the measurement of S-ASM activity, sodium acetate buffer was supplemented with 0.5 mM ZnCl2. After incubation for 0.5 to 4 h at 37 °C, the fluorescent product ceramide and uncleaved substrate were extracted by the addition of 250 μl chloroform/methanol (2:1, v/v). Following vortexing and centrifugation, the lipid phase was concentrated in a SpeedVac vacuum concentrator and spotted on silica gel 60 plates (Macherey-Nagel, Düren, Germany). Ceramide and sphingomyelin were separated by thin layer chromatography using chloroform/methanol (4:1, v/v) as a solvent and quantified on a Typhoon Trio scanner (GE Healthcare, 488 nm excitation and 520 nm emission wavelengths, 280 V, 100 μm resolution) with QuantityOne software (Bio-Rad, Munich, Germany). Measurements of all samples were performed in triplicates. For overexpression studies, each variant was transfected in triplicates within one experiment and all experiments were repeated at least once. The enzymatic activity of ASM was calculated as the rate of hydrolysis of sphingomyelin to ceramide per hour and per mg of protein in the cell lysate sample (pmol/mg/h).
Web Resources
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/.
Human Gene Mutation Database, http://www.hgmd.org/.
Results
High Frequency of ASM Sequence Variation c.1460C>T in the General Population
In a re-sequencing analysis of SMPD1 in 58 patients suffering from Major Depressive Disorder, we identified an unexpected high frequency of the SNP c.1460C>T (n = 3; allele frequency 2.6%). To determine whether the c.1460C>T variant is specific for patients suffering from MDD, we analysed another 998 DNA samples, 420 from patients with affective disorder and 578 from controls. Overall, nine (allele frequency 1.1%) and eight (0.69%) carriers of the T-allele were found among the patients and controls, respectively, indicating that the unexpected high frequency of the c.1460C>T variant is not associated with MDD.
Carriers of c.1460C>T Do Not Display Decreased Levels of ASM Activity
Next, we analysed the in vivo ASM activity of c.1460C>T carriers in a subgroup of the control sample (n = 156). Within this subgroup, two individuals carried the c.1460C>T variation (allele frequency 6.4%). Based on the analyses of He et al. (2003), we hypothesised that carriers of proposed NPD mutations should display lower S-ASM activity. The S-ASM activity in the individuals’ blood plasma was determined using a previously established enzymatic assay (Reichel et al. 2011). The S-ASM activities of all 156 individuals were used to establish a mean S-ASM activity level (196 ± 67 pmol/ml/h, n = 156). The S-ASM activity of carrier #1 was 80% of the mean S-ASM activity, and 42% activity was observed for carrier #2. We also determined the L-ASM activity in isolated lymphocytes, although the informative value of this activity level for the identification of NPD patients and carriers might be weak (Wenger et al. 1977). Carrier #1 exhibited an L-ASM activity of 62% and carrier #2 exhibited an L-ASM activity of 119% relative to the established mean L-ASM activity level (1.85 ± 0.62 pmol/mg/h, n = 156). Thus, only one of the two c.1460C>T carriers displayed a lower ASM activity, but this level was still above the level indicative of NPD carrier status as defined by the analysis of He et al. (2003). The other carrier exhibited ASM activity levels within a normal range.
ASM Variant p.A487V is Catalytically Active Upon Transient Overexpression
To further test the significance of the proposed NPD mutation c.1460C>T, we performed overexpression studies in cultured cells. As a control condition, we analysed the type B NPD mutation c.973C>G, which codes for the amino acid substitution p.P325A, in parallel. Variant cDNA plasmids were generated by site-directed mutagenesis and transiently overexpressed in HeLa and MDCK cells. Western blot analyses showed that the FLAG-tagged ASM variants were translated into proteins of the predicted sizes that were expressed to a similar extent as the wild-type ASM protein (Fig. 1). Overexpression of wild-type ASM in HeLa and MDCK cells increased the L-ASM activity in the cell lysates by 20-fold and 6-fold, respectively, and the S-ASM activity in the conditioned medium by 100-fold and 500-fold over endogenous levels, respectively. Overexpression of the p.P325A ASM variant did not increase the ASM activities above the endogenous levels, a result that is consistent with the notion that p.P325A is a loss-of-function mutation contributing to the development of NPD. In sharp contrast, overexpression of the ASM variant p.A487V resulted in ASM activities similar to that of wild-type ASM (Tables 1 and 2).
Fig. 1.
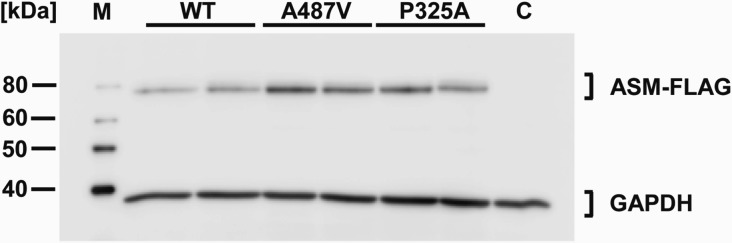
Western blot analysis of transiently expressed wild-type ASM and ASM variants. Cells transfected with constructs of wild-type ASM and the ASM variants p.A487V and p.P325A produced proteins with the predicted sizes. Lysates of transfected HeLa cells were analysed by Western blotting 48 h after transfection. ASM-FLAG variants were detected using an anti-FLAG antibody. Each second lane comprises the indicated variant with an additional common SNP for control reasons. The cloning vector served as a negative control (C). GAPDH was used as a loading control. M indicates the size marker.
Table 1.
L-ASM activities of transiently expressed ASM variants in HeLa and MDCK cells
| HeLa cell lysates | MDCK cell lysates | |||
|---|---|---|---|---|
| Construct | ASM activity (pmol/mg/h) |
% Wild-type activity |
ASM activity (pmol/mg/h) |
% Wild-type activity |
| Wild-type ASM | 24,579 | 100 | 35,679 | 100 |
| p.A487V | 26,014 | 106 | 34,229 | 95 |
| p.P325A | 915 | 1 | 5,778 | 1 |
| Mock control | 1,230 | 5,536 | ||
Table 2.
S-ASM activities of transiently expressed ASM variants in HeLa and MDCK cells
| HeLa cells | MDCK cells | |||
|---|---|---|---|---|
| Construct | ASM activity (pmol/ml/h) |
% Wild-type activity |
ASM activity (pmol/ml/h) |
% Wild-type activity |
| Wild-type ASM | 4,061 | 100 | 6,658 | 100 |
| p.A487V | 4,973 | 123 | 6,420 | 96 |
| p.P325A | 186 | 4 | 398 | 1 |
| Mock control | 40 | 0 | ||
Discussion
In this study, we analysed the properties of the proposed type B NPD mutation c.1460C>T (p.A487V) in detail. Contrary to expectations, the classification of variant c.1460C>T is debatable. Our analysis revealed an in vitro ASM activity that was comparable to the activity of wild-type ASM and almost normal S- and L-ASM activities in two carriers. In comparison, the proposed type B NPD missense mutation c.973C>G (p.P325A) (Simonaro et al. 2002), which was found in one patient suffering from MDD in our study, resulted in a severely reduced in vitro ASM activity. This result is in line with the classification as a deleterious mutation. It is still possible that c.1460C>T is associated with a defect that does not take effect under the chosen in vitro conditions, as reported for assays using the artificial sphingomyelinase substrate 2-N-(hexadecanoyl)-amino-4-nitrophenyl phosphorylcholine (Harzer et al. 2003). In addition, the high frequency of c.1460C>T carriers in our sample argues against the notion that c.1460C>T acts as an NPD mutation. However, we cannot exclude the possibility that the NPD allele frequency is underestimated in the general population. Consulting bioinformatic mutation prediction tools, our claim is further supported. The sequence variation c.1460C>T is supposed to be a polymorphism by Mutation Taster (Ramensky et al. 2002), neutral by SNAP (Bromberg and Rost 2007) and benign by PolyPhen-2 (score 0.014 out of 1.00) (Adzhubei et al. 2010).
What might be learned from these conflicting results? Considering the critical consequences that may arise from the classification, further analyses are warranted. It has to be taken into account that c.1460C>T was originally identified in an NPD patient as a proposed second defective allele. In some cases, c.1460C>T might be associated with another yet unknown polymorphism. Thus, either the effect of c.1460C>T could be modulated by a second site variant or the variant c.1460C>T could be acting as a surrogate marker for a more critical mutation in SMPD1. The existence of such a second marker is still speculative, and deeper investigations should be conducted to clarify this issue. The findings reported here, however, stress the importance of validating potential type A and B NPD mutations by various methodical approaches to ensure that they are deleterious mutations.
Acknowledgements
We thank Alice Konrad, Michaela Henkel, Sabine Müller, Stefan Hofmann and Michael Kathrein for excellent technical assistance. We are grateful to Sibylle Schwab for helpful discussions.
This work was supported by the German Federal Ministry for Education and Research BMBF (01GI0219; German Research Network on Depression and Suicidality) and by the Scholarship Programme ‘Equality for Women in Research and Teaching’, University Erlangen-Nuremberg (to CR).
Author Contributions
Conception and design: CR, MR, JK; Analysis of data: CR, JN, CM, PZ, MR; Interpretation of data: CR, JN, CM, PZ, MR; Drafting of article: CR, MR; Revising manuscript: JN, CM, PZ, UH, CH, RM, HJM, JK; Intellectual input: MA, UH, CH, RM, HJM, JK
Footnotes
According to Schuchman et al. (1991) numbered as A485V, here numbered according to GenBank Accession Number NM_000543.4 as A487V. The difference is due to the polymorphic region coding for the signal peptide of ASM.
Competing interests: None declared
References
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady RO, Kanfer JN, Mock MB, Fredrickson DS. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick disease. Proc Natl Acad Sci U S A. 1966;55(2):366–369. doi: 10.1073/pnas.55.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardis A, Zampieri S, Filocamo M, Burlina A, Bembi B, Pittis MG. Functional in vitro characterization of 14 SMPD1 mutations identified in Italian patients affected by Niemann Pick Type B disease. Hum Mutat. 2005;26(2):164. doi: 10.1002/humu.9353. [DOI] [PubMed] [Google Scholar]
- Desnick JP, Kim J, He X, Wasserstein MP, Simonaro CM, Schuchman EH. Identification and characterization of eight novel SMPD1 mutations causing types A and B Niemann-Pick disease. Mol Med. 2010;16(7–8):316–321. doi: 10.2119/molmed.2010.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt C, Schmolke M, Matzke A, et al. Polyethylenimine, a cost-effective transfection reagent. Signal Transduction. 2006;6:179–184. doi: 10.1002/sita.200500073. [DOI] [Google Scholar]
- Harzer K, Rolfs A, Bauer P, et al. Niemann-Pick disease type A and B are clinically but also enzymatically heterogeneous: pitfall in the laboratory diagnosis of sphingomyelinase deficiency associated with the mutation Q292 K. Neuropediatrics. 2003;34(6):301–306. doi: 10.1055/s-2003-44668. [DOI] [PubMed] [Google Scholar]
- He X, Chen F, Dagan A, Gatt S, Schuchman EH. A fluorescence-based, high-performance liquid chromatographic assay to determine acid sphingomyelinase activity and diagnose types A and B Niemann-Pick disease. Anal Biochem. 2003;314(1):116–120. doi: 10.1016/S0003-2697(02)00629-2. [DOI] [PubMed] [Google Scholar]
- Jones I, He X, Katouzian F, Darroch PI, Schuchman EH. Characterization of common SMPD1 mutations causing types A and B Niemann-Pick disease and generation of mutation-specific mouse models. Mol Genet Metab. 2008;95(3):152–162. doi: 10.1016/j.ymgme.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, Medlin A, Bleich S, et al. High activity of acid sphingomyelinase in major depression. J Neural Transm. 2005;112(11):1583–1590. doi: 10.1007/s00702-005-0374-5. [DOI] [PubMed] [Google Scholar]
- Pavlu-Pereira H, Asfaw B, Poupctova H, et al. Acid sphingomyelinase deficiency. Phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J Inherit Metab Dis. 2005;28(2):203–227. doi: 10.1007/s10545-005-5671-5. [DOI] [PubMed] [Google Scholar]
- Pinto R, Caseiro C, Lemos M, et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004;12(2):87–92. doi: 10.1038/sj.ejhg.5201044. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel M, Beck J, Mühle C, et al. Activity of secretory sphingomyelinase is increased in plasma of alcohol-dependent patients. Alcohol Clin Exp Res. 2011;35(10):1852–1859. doi: 10.1111/j.1530-0277.2011.01529.x. [DOI] [PubMed] [Google Scholar]
- Rhein C, Tripal P, Seebahn A et al (2012) Functional implications of novel human acid sphingomyelinase splice variants. PLoS ONE 7(4):e35467 [DOI] [PMC free article] [PubMed]
- Schuchman EH, Miranda SR. Niemann-Pick disease: mutation update, genotype/phenotype correlations, and prospects for genetic testing. Genet Test. 1997;1(1):13–19. doi: 10.1089/gte.1997.1.13. [DOI] [PubMed] [Google Scholar]
- Schuchman EH, Suchi M, Takahashi T, Sandhoff K, Desnick RJ. Human acid sphingomyelinase. Isolation, nucleotide sequence and expression of the full-length and alternatively spliced cDNAs. J Biol Chem. 1991;266(13):8531–8539. [PubMed] [Google Scholar]
- Simonaro CM, Desnick RJ, McGovern MM, Wasserstein MP, Schuchman EH. The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations. Am J Hum Genet. 2002;71(6):1413–1419. doi: 10.1086/345074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstein MP, Aron A, Brodie SE, Simonaro C, Desnick RJ, McGovern MM. Acid sphingomyelinase deficiency: prevalence and characterization of an intermediate phenotype of Niemann-Pick disease. J Pediatr. 2006;149(4):554–559. doi: 10.1016/j.jpeds.2006.06.034. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Barth G, Githens JH. Nine cases of sphingomyelin lipidosis, a new variant in Spanish-American Children. Juvenile variant of Niemann-Pick Disease with foamy and sea-blue histiocytes. Am J Dis Child. 1977;131(9):955–961. doi: 10.1001/archpedi.1977.02120220021002. [DOI] [PubMed] [Google Scholar]