

Abstract
Classical Menkes disease is a neurodegenerative disorder caused by mutations in the copper-transporting ATPase ATP7A gene which, when untreated, is usually fatal in early childhood. A mild form of Menkes disease was originally reported in 1981 and clinical progress of the patient at 10 years described subsequently. The causative mutation is c.4085C>T in exon 21, causing an alanine to valine substitution in the highly conserved TM7 domain at the C-terminal end of the Menkes protein. Here we report his status at 34 years of age. Intellectual impairment is mild. Ataxia has nearly resolved but motor retardation, dysarthria and an extreme slow speech rate remain. In contrast to patients with the occipital horn syndrome, there have been no connective tissue complications of his mild Menkes disease. He has been under long-term copper therapy for more than 30 years and he continues to enjoy a good quality of life.
Introduction
Menkes disease and the occipital horn syndrome are disorders at either ends of a spectrum caused by mutations in the copper-transporting ATPase ATP7A gene (OMIM #300011) (Kaler et al. 1994; Tumer et al. 1997). Additionally, specific mutations in ATP7A cause a distal motor neuropathy that is clinically distinct (Kennerson et al. 2010). Infant males with classical Menkes syndrome present in the first months of age with hypotonia, seizures and failure to thrive. Other manifestations include autonomic dysfunction, skin laxity, bladder diverticuli, pili torti hair changes and characteristic facies. Untreated boys with classical Menkes succumb in the first few years of life; however, early treatment with parenteral copper-histidinate may normalise the developmental outcomes in patients with residual enzyme activity (Kaler et al. 2008; Christodoulou et al. 1998). The occipital horn syndrome manifests with prominent connective tissue abnormalities as well as pathognomonic occipital exostoses; mild intellectual impairment may be seen (Tsukahara et al. 1994).
Between these two disorders lies the mild form of Menkes disease; the few described patients demonstrate developmental delay, variable connective tissue manifestations, pili torti and cerebellar ataxia but without seizures or childhood death (Gerdes et al. 1988; Proud et al. 1996; de Santos et al. 1984; Inagaki et al. 1988; Westman et al. 1988). Thus a clear distinction is drawn between classically affected patients with longer survival (including those treated with copper) and the mild Menkes patient.
Our patient was originally described by Procopis et al. in 1981 and his progress at 10 years of age was reported by David Danks (Procopis et al. 1981; Danks 1988). He presented with cerebellar ataxia, pili torti and vascular tortuosity. Follow-up demonstrated a predominantly motor delay with extreme dysarthria requiring a communication board until age 7. He commenced copper treatment at age 21 months and from 3 years has been on copper-histidinate injections. Here we describe his clinical status at the age of 34 years.
Case Report
When previously reported his major problems were cerebellar ataxia and dysarthria, both of which have improved, though not normalised, over time. He now mobilises independently without an ataxic gait and has a licence to drive a car. His movement is however not normal and is striking in the slowness with which he performs tasks. His speech remains extremely slow and dysarthric although the content is appropriate and his receptive understanding is not limited.
Formal intelligence testing has not been performed, but he is felt to function at the lowest end of normal. He underwent normal schooling, albeit with assistance from teachers, occupational therapists and speech pathologists. Long-term employment has been difficult to achieve, although he has worked periodically, predominantly due to his slowness of speech rather than intellectual impairment. Seizures have not occurred. Brain MRI was reported as normal at age 34.
Concurrent medical concerns have included nocturnal enuresis until the age of 20 years, subclinical hypothyroidism and frequent ear infections. Bladder ultrasound did not demonstrate diverticuli. He has long-standing and stable mild renal impairment with an estimated glomerular filtration rate of 57 ml/min/1.73 m2 (>60 ml/min/1.73 m2). There is stable microalbuminuria (urinary albumin:creatinine ratio 9.3 mg/mmol). Numerous investigations, but not including renal biopsy, have failed to demonstrate a cause and it is likely due to the damaging renal tubular effects of copper therapy (Lenartowicz et al. 2010).
His facial appearance in infancy had some features of the Menkes gestalt including pudgy cheeks and a carp-like upper lip. In adulthood these features have dissipated and he has a non-dysmorphic facial appearance. Pili torti was documented in previous reports; although his hair grows at a regular rate and he undergoes hair cuts every 6 weeks. Likewise his facial hair is normal and he sports a goatee beard (Fig. 1).
Fig. 1.
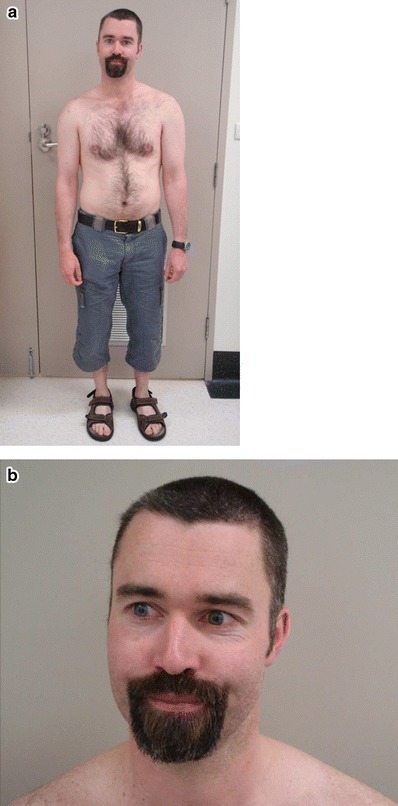
Appearance of the patient at age 34
There was no orthostatic hypotension and his plasma catecholamines were normal (adrenaline <0.3 nmol/l, 0.0–1.5; noradrenaline 1.6 nmol/l, 0.1–6.3). There was no history of diarrhoea or abnormal sweating.
Connective tissue appears largely normal, without extraordinary skin elasticity, varicose veins, joint hypermobility or bony abnormalities such as scoliosis, radial head deformity or dislocation. Lateral skull X-ray did not show wormian bones or occipital horns. Arteriography at 21 months demonstrated vascular tortuosity; however, cerebral magnetic resonance angiography at age 34 years demonstrated only mild tortuosity of the cavernous portions of the internal carotid arteries.
Copper-histidinate injections were maintained at 2.4 mg every second day from his mid-teenage years until the age of 32 years when they were changed to 3 mg every third day. The decrease in injection frequency was at the patient’s request as he found the injections increasingly difficult to comply with. Blood levels were monitored regularly via his local medical officer and most recently were slightly lower than usual with copper 9.1 umol/l (12–22) and caeruloplasmin 0.18 g/l (0.25–0.80).
The diagnostic biochemical parameters in blood, tissue and fibroblasts were reported in the original paper (Procopis et al. 1981), and since that time the family mutation in the ATP7A gene has been reported as c.4085C>T in exon 21, causing an alanine to valine substitution in the highly conserved TM7 domain at the C-terminal end of the Menkes protein that abrogates its copper-induced translocation to the plasma membrane (Ambrosini and Mercer 1999).
Discussion
In addition to our patient, a few other mild Menkes patients have been described and from this a phenotypic picture of variable intellectual impairment, ataxia and dysarthria, bladder diverticuli and vascular tortuosity emerges. Westman et al. described a 9 year old with near normal psychomotor abilities, bladder diverticuli and vascular tortuosity (Westman et al. 1988). Gerdes et al. described a 9 year old with borderline IQ and major problems of ataxia and bladder diverticuli (Gerdes et al. 1988). A more affected phenotype with prolonged survival comes from the family in which the responsible gene was cloned. These patients demonstrated various abnormalities across the spectrum of Menkes disease and Occipital Horn syndrome, with the 18 year old proband having mild intellectual disability, pili torti, bladder diverticuli, seizures, intracerebral haemorrhage, cutis laxa, vascular tortuosity, ataxia, occipital horns, scoliosis, pectus and multiple joint dislocations (Kaler et al. 1994; Proud et al. 1996).
Our patient tolerated copper-histidinate injections for more than 30 years and has demonstrated a slow improvement in his dysarthria and ataxia. Additionally, cerebral vascular tortuosity has lessened over time. Dysarthria and motor retardation remain his biggest challenges. There have not been long-term complications and in general he enjoys a good quality of life.
Author Contributions
Michel Tchan: Wrote the draft article. Guarantor.
Bridget Wilcken: Revised the article.
John Christodoulou: Revised the article.
All three authors have shared clinical care of this patient.
Competing Interest Statement
None reported.
Details of Funding
No funding was obtained for this study.
Ethics Approval and Patient Consent
Informed consent for publication of clinical information and photography was obtained from the patient and his mother.
Footnotes
Competing interests: None declared
References
- Alvarez de Santos M, Moreno P. Mild form of Menkes syndrome. Rev Invest Clin. 1984;36(2):151–154. [PubMed] [Google Scholar]
- Ambrosini L, Mercer JF. Defective copper-induced trafficking and localization of the Menkes protein in patients with mild and copper-treated classical Menkes disease. Hum Mol Genet. 1999;8(8):1547–1555. doi: 10.1093/hmg/8.8.1547. [DOI] [PubMed] [Google Scholar]
- Christodoulou J, et al. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients. Am J Med Genet. 1998;76(2):154–164. doi: 10.1002/(SICI)1096-8628(19980305)76:2<154::AID-AJMG9>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Danks DM. The mild form of Menkes disease: progress report on the original case. Am J Med Genet. 1988;30(3):859–864. doi: 10.1002/ajmg.1320300325. [DOI] [PubMed] [Google Scholar]
- Gerdes AM, et al. Variability in clinical expression of Menkes syndrome. Eur J Pediatr. 1988;148(2):132–135. doi: 10.1007/BF00445920. [DOI] [PubMed] [Google Scholar]
- Inagaki M, et al. Atypical form of Menkes kinky hair disease with mitochondrial NADH-CoQ reductase deficiency. Neuropediatrics. 1988;19(1):52–55. doi: 10.1055/s-2008-1052402. [DOI] [PubMed] [Google Scholar]
- Kaler SG, et al. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet. 1994;8(2):195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- Kaler SG, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358(6):605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerson ML, et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010;86(3):343–352. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenartowicz M, et al. Effects of copper supplementation on the structure and content of elements in kidneys of mosaic mutant mice. Biol Trace Elem Res. 2010;136(2):204–220. doi: 10.1007/s12011-009-8533-4. [DOI] [PubMed] [Google Scholar]
- Procopis P, Camakaris J, Danks DM. A mild form of Menkes steely hair syndrome. J Pediatr. 1981;98(1):97–99. doi: 10.1016/S0022-3476(81)80551-3. [DOI] [PubMed] [Google Scholar]
- Proud VK, et al. Distinctive Menkes disease variant with occipital horns: delineation of natural history and clinical phenotype. Am J Med Genet. 1996;65(1):44–51. doi: 10.1002/(SICI)1096-8628(19961002)65:1<44::AID-AJMG7>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Tsukahara M, et al. Occipital horn syndrome: report of a patient and review of the literature. Clin Genet. 1994;45(1):32–35. doi: 10.1111/j.1399-0004.1994.tb03986.x. [DOI] [PubMed] [Google Scholar]
- Tumer Z, et al. Identification of point mutations in 41 unrelated patients affected with Menkes disease. Am J Hum Genet. 1997;60(1):63–71. [PMC free article] [PubMed] [Google Scholar]
- Westman JA, et al. Atypical Menkes steely hair disease. Am J Med Genet. 1988;30(3):853–858. doi: 10.1002/ajmg.1320300324. [DOI] [PubMed] [Google Scholar]