

Abstract
Pompe disease, especially in its infantile form, is a fatal disease. Most of the patients with this disease synthesize a nonfunctional form of the enzyme alpha glucosidase (GAA), the deficient enzyme in this disease. Patients producing some amount of this protein are labeled as cross-reactive immunologic material (CRIM)-positive. Few of them are unable to synthesize it and are labeled CRIM-negative. The clinical course of the disease has changed with the advent of enzyme replacement therapy (ERT) with recombinant alpha glucosidase enzyme (rhGAA). However, CRIM-negative patients have always been known to have poor outcome on ERT due to the development of anti-rhGAA antibodies in their bodies that neutralizes ERT efficacy. Here, we describe two CRIM-negative siblings on rhGAA ERT with unusually low anti-rhGAA antibody titer and good clinical outcome. Up to our current knowledge, this is the first report that describes such a good response to ERT in CRIM-negative patients.
Introduction
Pompe disease (glycogen storage disease type II) is an autosomal recessive lysosomal storage disease caused by deficiency of acid α-glucosidase (GAA). As a consequence, its substrate glycogen accumulates in cardiac, skeletal, and smooth muscle tissues. The infantile form, which is the most severe phenotype, presents clinically with cardiomyopathy, hypotonia, generalized muscle weakness, feeding difficulties, failure to thrive, and respiratory insufficiency.
GAA protein is called cross-reactive immunologic material (CRIM) because anti-GAA antibodies recognize it on Western blot analysis. Most Pompe patients still have some residual GAA protein production and are labeled as CRIM positive while those with total absence of that protein are labeled CRIM negative (Amalfitano et al. 2001). Without treatment, death typically occurs with the first 1 to 2 years secondary to cardiorespiratory failure (Van den Hout et al. 2003). Enzyme replacement therapy (ERT) with recombinant acid α-glucosidase (rhGAA) has been shown to prolong these patients’ lives (Kishnani et al. 2007, 2009; Nicolino et al. 2009). However, the response of these patients to rhGAA treatment depends on their CRIM status. CRIM-negative infants generally have a poor prognosis, where they have been found to have an initial improvement followed by a clinical deterioration. Their deterioration has been attributed to the high levels of anti-rhGAA IgG antibodies their bodies develop against the recombinant enzyme. Conversely, CRIM-positive infants have a more favorable prognosis and their anti-rhGAA antibodies are not as significantly elevated as CRIM-negative ones (Kishnani et al. 2010; Banugaria et al. 2011).
In this case report, we describe two siblings, brother and sister, who are CRIM negative on a relatively long GAA ERT with good outcome.
Patients and Methods
The patients are two African American CRIM-negative siblings. The first is a 4.5-year-old boy while the second is 2-year-old girl.
CRIM status was determined as described previously (Kishnani et al. 2006). Anti-rhGAA IgG antibody titer measurements and CRIM status were performed at Genzyme Corporation (Cambridge, MA). Leukocyte GAA activity assays were performed at Emory University Biochemical Genetics Laboratory. GAA mutation analysis was performed at Emory University Molecular Genetics Laboratory.
Results
The boy was a product of full-term pregnancy delivered via cesarean section secondary to abnormal heart rate deceleration. Soon after delivery, he was noted to be mildly cyanotic and was given a diagnosis of transient tachypnea of the newborn (TTN). A chest x-ray showed left ventricular enlargement. A follow-up echocardiogram showed hypertrophic obstructive cardiomyopathy with mild left ventricular outflow tract obstruction and mild mitral regurgitation. He was started on propranolol, which lead to improvement in the mitral regurgitation and left ventricular outflow tract gradient. He was discharge on propranolol 2 mg every 6 h to be followed bimonthly by the cardiologist. Because of the progressive worsening of his cardiomyopathy, his cardiologist referred him to our genetics department to investigate for a possible genetic cause of his condition, specifically to rule out Pompe disease. When he was first seen in our genetics clinic, he was 6 months old and his investigations showed total creatine kinase (CPK) level of 2902 IU/L (normal: 60–305 IU/L) and CK MB fraction of 19.9 ng/mL (normal range: 0.0–3.3 ng/mL). Pompe disease was suspected and diagnosed on the basis of decreased leukocyte GAA activity (0.3 nmoles/mg protein/h; normal: 6.01–37 nmoles/mg protein/h) and confirmed by mutation analysis that showed a deletion mutation in one allele (IVS2 +2_ +5 delTGGG deletion in intron 2) and an insertion mutation (c.1650_1651 dupG in exon 12) in the second. Western blot testing on a skin fibroblast sample showed him to be CRIM-negative.
Since his diagnosis was confirmed, he has been receiving 20 mg/kg rhGAA every 2 weeks, now for a total of 4 years on treatment. His clinical course was complicated by one hospital admission at the age of 1.5 years for pneumonia for which he was intubated, but is now completely recovered. Since that hospitalization, he has been followed annually with a pulmonologist who prescribed him Levalbuterol and Albuterol nebulizers. He has regular follow up with his cardiologist. He still has mild intraventricular conductive delay and mild hypertrophy. His echocardiogram showed significant improvement since he was started on rhGAA ERT; he currently has a ventricular septum of 7 mm, which has been stable for the last 12 months, and is significantly decreased compared to his highest measurement of 18 mm just before the start of his ERT (Fig. 1a and b). His anti-rhGAA antibody titers over the course of his ERT are summarized in Fig. 2. During the course of his treatment, he never had any infusion-related events even at the time of his highest anti-rhGAA antibody titer.
Fig. 1.
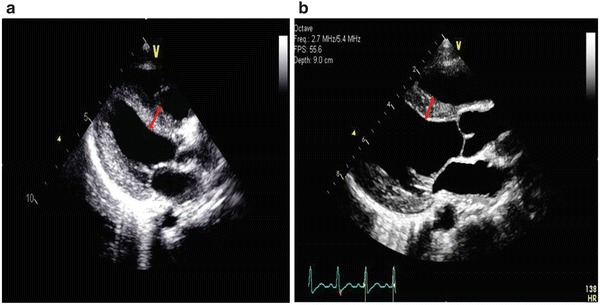
Echocardiogram images showing the first patient’s interventricular septum thickness (red bidirectional arrow) before (a) and after (b) ERT
Fig. 2.
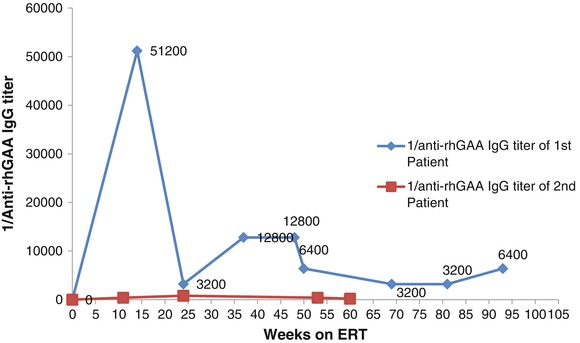
Patients’ anti-rhGAA antibody titers over their corresponding duration of treatment
He has mild developmental delay. He walked at the age of 17 months. He receives physiotherapy and can run and walk up and down stairs. He wears ankle-foot orthotic braces now on a daily basis due to a development of foot drop in the last year. He has been found to have low vitamin D level, low borderline calcium, and elevated parathyroid hormone. He is now on supplemental vitamin D and calcium. His receptive language is normal but expressive skills are delayed for which he is receiving speech therapy. He has passed all his hearing tests so far.
Based on the positive family history, his sister’s diagnosis was reached prenatally via mutational analysis on amniocyte-isolated DNA where she was found to have the same mutations as her brother. Her leukocyte GAA activity was 1.02 nmoles/mg protein/h (normal: 6.01–37 nmoles/mg protein/h). At the age of 2 days, her CPK level was found to be 1350 IU/L. Her echocardiogram was consistent with severe obstructive cardiomyopathy for which she was transferred to pediatric intensive care unit. She has been receiving 20 mg/kg rhGAA every 2 weeks since the age of 6 days, now on ERT for over 2 years. She still has mild intraventricular conductive delay and T-wave inversion in the anterolateral leads with features of ventricular strain. Her last echocardiogram showed a ventricular septum thickness of 5 mm, which has been persistently decreasing from her highest, i.e., 11 mm at birth (Fig. 3a and b). She passed all her hearing tests so far and has been developing normally with no hospitalization or decompensation. Her anti-rhGAA antibody titers over the course of her ERT are summarized in Fig. 2. Unlike her brother, she had multiple infusion-related reactions in the forms of hives that respond very well to 11 mg oral dose of diphenhydramine hydrochloride and infusion time extension. None of these reactions were life threatening or severe enough to stop the infusion or decrease its frequency.
Fig. 3.
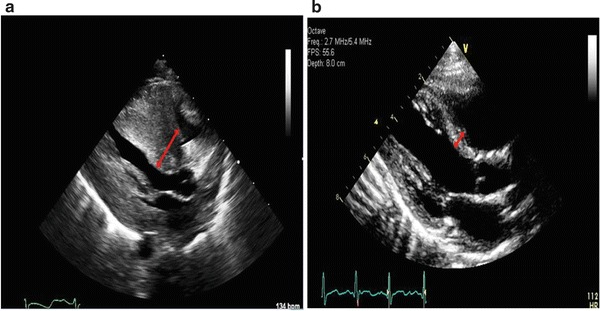
Echocardiogram images showing the second patient’s interventricular septum thickness (red bidirectional arrow) before (a) and after (b) ERT
Discussion
Since its approval for commercial use in 2006, ERT with rhGAA has yielded significant change in the course of infantile Pompe disease. It has been found, particularly in CRIM-positive patients, to reverse cardiomyopathy, delay or stabilize motor disease progression characterizing this disease, and prolong ventilator-free survival (Banugaria et al. 2011). Because of the high and sustained titers of neutralizing anti rhGAA antibodies produced in their bodies, CRIM-negative patients are known to benefit the least from such therapy and they are either ventilator dependent or deceased by age 27.1 months (Abbott et al. 2011). The longest CRIM-negative ventilator-free survival was described in a single case report (Rohrbach et al. 2010) in which the patient was treated with omalizumab (anti-IgE mAb) for IgE-mediated anaphylactic episode related to ERT. The elder CRIM-negative patient in our case report has so far reached the age of 54 months, which is the longest ever ventilator-free survival reported for a CRIM-negative patient. In addition, he completed this ventilator-free period of time without having any immunomodulatory intervention.
The nature of the mutation in these two siblings is a candidate reason behind their favorable outcome to enzyme treatment compared to other CRIM-negative patients. Both mutations have recently been reported and associated with CRIM-negative status (Bali et al. 2012). To our knowledge, no study to correlate the type of mutation in CRIM-negative patients with their response to rhGAA ERT was published. In these two siblings, it is possible that the product of the first mutation, a splice mutation, might produce a quantity of the enzyme that is too small to be detected by western blot and yet enough to desensitize the patient’s immune system so that it would not produce high titer of anti-rhGAA antibodies. Since the second mutation is a frame shift mutation with nonesense mutation 85 base pairs downstream, we do not expect a protein to be produced by this mutation especially when we take into consideration the nonesense-mediated messenger RNA decay that occurs with such mutations.
The fact that the second sibling started her enzyme therapy earlier than the first one, explains her lower anti-rhGAA antibody titers compared to her brother who carries the same mutations and points toward the importance of early enzyme treatment.
This case report does not only describe a new pattern of immunological response to rhGAA ERT but emphasizes the fact that our understanding of that immunological process is limited. It also proves that CRIM status is not the sole immunological determinant of Pompe patients’ prognosis on such treatment and reinforces the need to study the genotype-phenotype correlation between CRIM-negative mutations and the response to enzyme treatment.
With the availability of immunomodulatory regimens (Joseph et al. 2008; Mendelsohn et al. 2009; Rohrbach et al. 2010; Messinger et al. 2012) to combat immunological reaction against rhGAA that CRIM-negative patients usually develop, it is clear from these two cases that not all CRIM-negative patients need them; relying solely on CRIM status to decide who should receive such immunomodulatory treatment might subject some CRIM-negative patients to unnecessary immunosuppression. This issue holds further importance as Pompe disease is being proposed to be included on the newborn screening panel. If added, consensus guidelines will be needed to address timing of ERT initiation in CRIM-negative patients and if immunomodulatory regimen should be used.
Acknowledgments
We thank Joe Kreeger, echocardiography laboratory supervisor, Children's Healthcare of Atlanta, for encrypting the echocardiograms published in this study.
Abbreviations
- CRIM
Cross reactive immunologic material
- ERT
Enzyme replacement therapy
- GAA
Acid alpha glucosidase
- rhGAA
Recombinant human acid alpha glucosidase
Synopsis
Some CRIM-negative patients can have good outcome on enzyme replacement therapy
Authors’ Contribution
Hamoud H. Al Khallaf contributed to this work by gathering some data, drawing a figure, and participating in writing the manuscript.
Jennifer Propst contributed to this work by summarizing patients’ clinical data and revising the manuscript.
Eleanor Botha helped in summarizing patients’ clinical data and revising the manuscript.
Serge Geffrard performed echocardiogram for the patients and revised the manuscript.
M. Ali Pervaiz planned the study, obtained the clinical data from the patients, and edited the entire manuscript.
Funding
None.
Ethics Approval
Was not required.
Patients’ Consents
Was obtained from the mother of the two subjects.
Conflict of Interest
None.
Footnotes
Competing interests: None declared
Contributor Information
Hamoud H. Al Khallaf, Email: hamoud.h.al.khallaf@emory.edu.
Jennifer Propst, Email: jennifer.propst@emoryhealthcare.org.
Serge Geffrard, Email: sergegeffrard@yahoo.com.
Eleanor Botha, Email: eleanor.botha@emoryhealthcare.org.
M. Ali Pervaiz, Email: alipervaiz@emory.edu.
References
- Abbott MA, Prater SN, Banugaria SG, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab. 2011;104:583–586. doi: 10.1016/j.ymgme.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alphaglucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. [PubMed] [Google Scholar]
- Bali DS, Goldstein JL, Banugaria S, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease usingGAA mutations: Lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet. 2012;160:40–49. doi: 10.1002/ajmg.c.31319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph A, Munroe K, Housman M, Garman R, Richards S. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clin Exp Immunol. 2008;152:138–146. doi: 10.1111/j.1365-2249.2008.03602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66:329–335. doi: 10.1203/PDR.0b013e3181b24e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, et al. Crossreactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med. 2009;360:194–195. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]
- Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM –negative infantile Pompe disease. Genet Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- Rohrbach M, Klein A, Kohli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- Van den Hout HM, Hop W, Van Diggelen OP, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]