

Abstract
Anderson-Fabry disease (AFD) is a multiorgan X-linked lysosomal storage disease that particularly affects the heart, kidneys, and cerebrovascular system. Current treatment is enzyme replacement therapy (ERT) with agalsidase beta (Fabrazyme®, Genzyme Corporation, Cambridge, MA, USA) or agalsidase alfa (Replagal®, Shire Human Genetic Therapies AB, Lund, Sweden). It was recommended that patients switch to agalsidase alfa due to a manufacturing shortage of agalsidase beta beginning in June 2009. This study assessed the effect of switching to agalsidase alfa on clinical outcomes in patients with AFD previously treated with agalsidase beta. Ten patients (seven male, three female) with genetically confirmed AFD and at least 48 months’ continuous data collected during treatment with agalsidase beta 1 mg/kg every other week were switched to agalsidase alfa 0.2 mg/kg every other week for at least 20 months, with prospective clinical evaluations every 6 months. Pre-switch data was collected retrospectively from patient charts. Cardiac functional parameters were assessed using magnetic resonance imaging. Results showed that renal function was normal (estimated glomerular filtration rate ≥90 mL/min/1.73 m2) in 8 of 10 patients prior to agalsidase alfa and generally remained stable after the switch. Cardiac mass decreased significantly (p < 0.05 vs pre-ERT) after agalsidase beta and remained unchanged after switching to agalsidase alfa. Symptoms of pain and health status scores did not deteriorate during agalsidase alfa therapy. Adverse events were mostly mild and infusion related. In conclusion, switching to agalsidase alfa was relatively well tolerated and associated with stable clinical status and preserved renal and cardiac function.
Introduction
Anderson-Fabry disease is an X-linked lysosomal storage disorder caused by a deficiency of the hydrolytic enzyme α-galactosidase A (α-Gal A). Consequently, globotriaosylceramide (Gb3) accumulates in cells and tissues of the body resulting in a multisystem pathology (Brady et al. 1967; Desnick et al. 2001), affecting the skin, nervous and cerebrovascular systems, kidneys, and heart, and is associated with a reduced life expectancy (Zarate and Hopkin 2008). Renal involvement is progressive, eventually leading to end-stage renal disease (ESRD) by the fourth and fifth decades of life if untreated; prior to the advent of dialysis and renal transplantation, ESRD was the leading cause of death (Branton et al. 2002). Cardiac manifestations include left ventricular (LV) hypertrophy, valvular disease, conduction abnormalities leading to arrhythmias, congestive heart failure, and coronary artery disease (Weidemann et al. 2005; Linhart and Elliott 2007; Kampmann et al. 2008; Linhart 2008; Morrissey et al. 2011).
Two recombinant enzyme formulations for enzyme replacement therapy (ERT) of Anderson-Fabry disease have been commercially available in Europe for almost 10 years: agalsidase alfa (Replagal®, Shire Human Genetic Therapies AB, Danderyd, Sweden) and agalsidase beta (Fabrazyme®, Genzyme Corporation, Cambridge, MA, USA) (Smid et al. 2011). Numerous clinical trials, observational studies, and registry data have provided some evidence for the safety and efficacy of ERT in improving disease symptoms, cardiac mass, renal function, and quality of life (Eng et al. 2001; Schiffmann et al. 2001; Weidemann et al. 2003; Wilcox et al. 2004; Banikazemi et al. 2007; Hughes et al. 2008; Koskenvuo et al. 2008; Imbriaco et al. 2009; Mehta et al. 2009; Feriozzi et al. 2012). To date, there have been limited comparisons of the two agents, from which no firm conclusion can be drawn regarding their relative efficacy and safety (Lidove et al. 2010).
As of June 2009, viral contamination in the agalsidase beta production facility resulted in a worldwide supply shortage of agalsidase beta (European Medicines Agency 2009). It was initially recommended by the European Medicines Agency (EMA) that priority patients continue on a reduced dosage of agalsidase beta (European Medicines Agency 2009) but this appeared to be associated with an increase in the adverse event rate, prompting the EMA to revise their recommendation about a year later to treatment with either the full dose of agalsidase beta (1 mg/kg every 2 weeks) or agalsidase alfa (0.2 mg/kg every 2 weeks) (European Medicines Agency 2010a, b). An expert consensus guidelines published in 2011 recommended a similar approach (Linthorst et al. 2011). Data on the effect of switching between the two therapies are limited to one observational study in 11 Japanese patients (Tsuboi and Yamamoto 2012) and a subgroup of 20 patients in a retrospective cohort Dutch study (Smid et al. 2011). Both reported clinical outcomes, including effects on cardiac abnormalities, but neither assessed cardiac effects via cardiac magnetic resonance imaging (cMRI).
We aimed to assess the effect of switching from agalsidase beta to agalsidase alfa on renal function, cardiac functional parameters (assessed via cMRI), health status, pain, and adverse events in patients with Anderson-Fabry disease by comparing retrospective data during treatment with agalsidase beta with prospective data during 20 months’ treatment with agalsidase alfa.
Materials and Methods
Study Design
This was a single-center observational study conducted at the University Hospital Federico II, Italy, of patients with Anderson-Fabry disease who switched from treatment with agalsidase beta to agalsidase alfa. Data for the period during which patients received agalsidase beta was gathered retrospectively. Data collected at the time patients switched to agalsidase alfa and during agalsidase alfa treatment was gathered prospectively.
The study was conducted in accordance with the Declaration of Helsinki and was approved by the institutional ethics committee. All patients switched to agalsidase alfa and included in the final study population provided written informed consent.
Patients
Patients eligible for study inclusion were those with a genetically confirmed diagnosis of Anderson-Fabry disease, at least 48 months of continuous data during treatment with agalsidase beta 1 mg/kg (i.e., prior to switching), those switched to agalsidase alfa after the start of the agalsidase beta shortage, and patients who provided written informed consent. Patients excluded from the study included those who had received agalsidase beta at other than the recommended dose for >12 months prior to the study or who had used any investigational drug within 30 days prior to study entry.
Treatments
Patients receiving agalsidase beta at a dosage of 1 mg/kg every other week for a minimum of 48 months were switched without any crossover period or interval to agalsidase alfa 0.2 mg/kg every other week, given according to the manufacturer’s prescribing information. Patients received agalsidase alfa for a minimum of 20 months. Pretreatment with an antihistamine and/or ibuprofen was allowed to minimize infusion-associated reactions (IAR). Concomitant treatments such as antihypertensive agents were allowed.
Data Collection and Study End Points
Study endpoints included renal function, selected cardiac parameters, pain symptoms, and patient health status. The following clinical evaluations were performed at baseline, after 48 months of ERT with agalsidase beta, and again after 20 months ERT with agalsidase alfa: (1) physical examination, routine blood chemistries, hematology, and urinalysis; (2) serum creatinine levels and the mean estimated glomerular filtration rate (GFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation (Levey et al. 1999) on the basis of patients’ age, race, sex, and serum-creatinine values; (3) proteinuria (estimated via the urine protein to creatinine ratio); (4) 12-lead electrocardiography (ECG); (5) cMRI was used to determine LV mass, LV wall thickness, and LV ejection fraction (see separate section for cMRI Techniques and Analysis); (6) health status, evaluated with the Short Form-36 (SF-36) Health Status Survey (Ware 1997); and (7) pain score, evaluated with the short form McGill pain questionnaire (Melzack 1987). Use of concomitant medications, including pain medication and antihypertensive agents was recorded. In addition, α-Gal A enzyme activity was assessed at diagnosis (Desnick et al. 1973).
Data collected during treatment with agalsidase beta was obtained retrospectively from patient medical charts for the 48 months prior to the treatment switch. Retrospective data included all endpoints as described above for the prospective part of the study and, in addition, the results of a physical examination and cMRI prior to initiation of agalsidase beta.
cMRI Technique and Analysis
cMRI assessments were performed pre-ERT, after 48 months’ agalsidase beta (before switching), and after 20 months’ agalsidase alfa, using a 1.5 Tesla MRI system (Gyroscan Intera, Philips Medical System, Best, the Netherlands) equipped with high-performance gradients (maximum gradient amplitude 30 mT/m, maximum slew rate 150 mT/m/ms). Images were acquired with a 5-element cardiac phased-array coil using a vector cardiographic method for ECG gating and respiratory gating. After performing a survey scan, LV long axis and 4-chamber horizontal long axis images were acquired using a breath-holding 2D balanced turbo field echo multiphase-multislice sequence (TR/effective TE, 2.8/1.4; matrix, 160X256; slice thickness, 10-mm; flip angle, 50°); subsequently, biventricular short-axis images were obtained using 9–10 slices covering the left ventricle from the apex to the base for evaluation of LV mass. The total acquisition time ranged between 25 and 30 min for each assessment.
Post-processing was performed on a dedicated workstation (Viewforum, Philips Medical System, Best, the Netherlands). LV wall thickness was measured at the level of the mid-septum. Analysis of LV mass was performed choosing the slice with the greatest cardiac diameter of the 2D-balanced turbo field echo multiphase-multislice acquisition in the biventricular short axis; subsequently, the endocardial and the epicardial borders were manually traced, carefully including the papillary muscles, on each end-diastolic and end-systolic frame for each of the 9–10 slices.
The evaluation of late gadolinium enhancement (LGE), looking for areas of myocardial fibrosis, although not a goal of this study, was assessed and only in two cases were these additional MR sequences observed. In particular, a commercially available gadolinium-based contrast agent, gadodiamide (gadopentetate dimeglumine – Magnevist, Schering AG, Berlin, Germany; 0.15 mmol/kg), was injected intravenously at a dose of 0.1 mmol/kg of body weight, and 10–15 min after the injection, a segmented inversion-recovery fast-gradient echo sequence was acquired. LGE images were acquired in multiple short-axis views identical to those obtained for cine cardiac MRI. Finally, a visual estimation of LGE extent in LV horizontal, vertical long-axis, and short axis views was subsequently performed.
Safety
The safety of agalsidase alfa was assessed by recording all adverse events (AEs), which were assessed for their severity and relationship to the study drug.
Statistical Analysis
Data are mean ± standard deviation (SD) unless specified otherwise. Differences in LV parameters between pre-ERT and agalsidase beta treatment, and between agalsidase beta and agalsidase alfa treatment, were analyzed using a Student t-test for paired observations. Value of p < 0.05 was considered statistically significant.
Results
Patients
Of the 14 patients with Anderson-Fabry disease receiving agalsidase beta at our institution, 10 agreed to switch to agalsidase alfa, 2 patients opted for the reduced agalsidase beta dosage (0.3 mg/kg every other week) and 1 discontinued ERT. Ten patients (mean ± SD age 44 ± 5 years) with genetically confirmed Anderson-Fabry disease met study inclusion criteria and were enrolled (seven male, three female) into the prospective part of the study. A causal mutation/deletion was identified in all patients included in this study.
Renal Function
Renal function, assessed by serum creatinine levels and eGFR, remained stable in both male and female patients 24 months after switching treatment (Table 1). Individual eGFR values confirmed this finding, with no patient demonstrating clinically significant changes in eGFR after the ERT switch (Table 2). At entry to the evaluation, eGFR was normal (eGFR ≥90 ml/min/1.73 m2) in 8 of the 10 patients, and remained normal during the 20-month follow-up. Two patients had elevated serum creatinine levels at the start of the switch but these levels did not worsen with agalsidase alfa treatment (data not shown). The median urinary protein to creatinine ratio was 0.96 (in mg protein/mMol creatinine) pre-ERT, 0.36 after 48 months’ agalsidase beta therapy (before switching to agalsidase alfa), and 0.38 after 20 months’ agalsidase alfa therapy, indicating a stabilization in urine protein excretion. Individual patient data showed that those patients who experienced an increase in urine protein excretion had relatively high proteinuria at baseline (data not shown).
Table 1.
Selected study endpoints at pre-ERT, after 48 months’ treatment with agalsidase beta and 20 months’ agalsidase alfa. Data are mean ± SD values unless stated otherwise
| Pre-ERT | After 48 months’ agalsidase beta | After 20 months’ agalsidase alfa | |
|---|---|---|---|
| Renal function | |||
| eGFR (L/min/1.73 m2) | 92.4 ± 13.1 | 91.3 ± 14.9 | 90.3 ± 17.5 |
| Median (range) eGFR (L/min/1.73 m2) | 98 (67–100) | 97.5 (60–100) | 96 (60–100) |
| Proteinuria | |||
| Urinary protein to creatinine ratio | 0.96 ± 0.29 | 0.36 ± 0.14 | 0.38 ± 0.07 |
| Cardiac parameters | |||
| LV mass (g/m2) | 106 ± 32 | 73 ± 24a,b | 70 ± 24 |
| LV wall thickness (mm) | 16 ± 4 | 13 ± 4a,b | 13 ± 3 |
| LVEFc (%) | 63 ± 4 | 65 ± 6 | 64 ± 6 |
eGFR estimated glomerular filtration rate; LV left ventricular; LVEF c LV ejection fraction, corrected; SD standard deviation
a p < 0.05 vs baseline
b p > 0.05 vs agalsidase alfa
c p > 0.05 baseline vs agalsidase beta vs agalsidase alfa
Table 2.
Individual patient data for estimated glomerular filtration rate (GFR; L/min/1.73 m2) at baseline and after 48 months’ treatment with agalsidase beta and after 20 months’ agalsidase alfa treatment
| GFR, L/min/1.73 m2 | |||
|---|---|---|---|
| Patient | Pre-ERT | After 48 months’ agalsidase beta | After 20 months’ agalsidase alfa |
| 1 | 100 | 100 | 98 |
| 2 | 68 | 68 | 65 |
| 3 | 67 | 60 | 60 |
| 4 | 100 | 100 | 100 |
| 5 | 98 | 98 | 96 |
| 6 | 98 | 96 | 95 |
| 7 | 97 | 97 | 97 |
| 8 | 98 | 96 | 96 |
| 9 | 98 | 98 | 96 |
| 10 | 100 | 100 | 100 |
Cardiac Functional Parameters
Cardiac function (assessed by LV ejection fragment [LVEF]) remained unchanged after a switch from agalsidase beta to agalsidase alfa (Table 1); pre-ERT LVEF values did not change significantly after agalsidase beta or agalsidase alfa therapy. LV mass index and LV wall thickness decreased significantly (p < 0.05) from pre-ERT values after 48 months’ treatment with agalsidase beta as shown in Table 1. These reductions were maintained when patients were switched to agalsidase alfa, as there were no significant differences in these parameters after 20 months’ treatment with agalsidase alfa compared with values after 48 months’ agalsidase beta treatment. Individual changes in left ventricular functional parameters measured at baseline after 48 months of therapy with agalsidase beta and after 20 ± 3 months of agalsidase alfa therapy are shown in Table 3.
Table 3.
Left ventricular functional parameters measured at baseline (1), after 48 months of therapy with agalsidase beta (2) and after 20 ± 3 months of therapy with agalsidase alfa (3)
| Pts (n = 10) | Age/Gender | LVMI (g/m2) | LVMI (g/m2) | LVMI (g/m2) | LVWT (mm) | LVWT (mm) | LVWT (mm) |
|---|---|---|---|---|---|---|---|
| Time point | 1 | 2 | 3 | 1 | 2 | 3 | |
| 1 | 56/M | 167 | 113 | 92 | 21 | 15 | 15 |
| 2 | 43/M | 82 | 70 | 64 | 15 | 15 | 13 |
| 3 | 44/M | 77 | 55 | 53 | 15 | 10 | 12 |
| 4 | 43/F | 147 | 111 | 116 | 22 | 22 | 20 |
| 5 | 38/F | 93 | 62 | 64 | 15 | 11 | 13 |
| 6 | 40/M | 66 | 71 | 54 | 13 | 9 | 10 |
| 7 | 41/M | 98 | 75 | 64 | 12 | 10 | 10 |
| 8 | 50/M | 121 | 50 | 52 | 15 | 14 | 13 |
| 9 | 40/F | 85 | 41 | 42 | 14 | 12 | 11 |
| 10 | 48/M | 124 | 86 | 96 | 14 | 12 | 15 |
LVMI left ventricular mass index, LVWT left ventricular wall thickness
No significant correlation was observed between the age of the patients and the variation in LV functional parameters (p > 0.05) with agalsidase beta or with agalsidase alfa treatment.
Five male patients showed signs of LV hypertrophy (LVH) before starting ERT with agalsidase beta. LVH improved after 48 months’ therapy with agalsidase beta, and this remained at a stable level after 20 months’ agalsidase alfa ERT. Figure 1 shows no changes in LV wall thickness and LV mass, between study 2 and 3 in a 50-year-old male patient with Anderson-Fabry Disease.
Fig. 1.
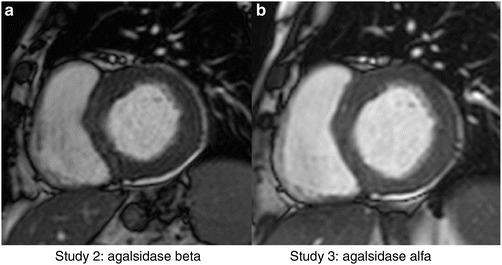
Left ventricular short axis view in a 50-year-old male with Anderson-Fabry disease showing no changes in LV mass index and LV wall thickness between 48 months’ ERT with agalsidase beta (50 g/m2, 14 mm) and after 20 months’ ERT with agalsidase alfa (52 g/m2, 14 mm)
Quality of Life
For most components of the SF-36 questionnaire, there were no differences in mean scores after agalsidase alfa treatment compared with the pre-switch period (data not shown). No variations from pre-treatment through month 20 were seen for the components of Physical Functioning, Role Emotional, Body Pain, and Standardized Physical Component Scale; only small improvements were observed for the other components, although none reached statistical significance (data not included).
Pain Symptoms
Mean pain scores improved during agalsidase beta ERT and then remained stable (i.e., no worsening) during treatment with agalsidase alfa (data not inclued). Of the 10 patients, 8 had no pain at switching and remained pain free during treatment with agalsidase alfa; 2 patients were experiencing pain at the time of switching, but then improved slightly during the agalsidase alfa ERT period.
Tolerability
There were no reports of treatment-related atrial fibrillation, ventricular premature beats, tachyarrhythmia, cardiac failure, or other cardiac adverse events within 24 h of administration of ERT for every infusion given during the 20 months’ agalsidase alfa ERT period. Six patients experienced at least one AE during treatment with agalsidase alfa. Most AEs were mild in nature and unrelated to treatment. The most common treatment-related AEs were IAR, consisting of rigors, temperature change sensations, fever, nausea, headache, vomiting, flushing, rhinitis, pruritus, and somnolence. Most IARs were assessed as mild, and the total number of patients who experienced an IAR markedly decreased over time (data not shown). There were no abnormal laboratory test values. No major AE were observed during treatment with agalsidase beta.
Discussion
Renal and cardiac function, pain symptoms and health status were largely unchanged when 10 patients with Anderson-Fabry disease switched from ERT with agalsidase beta to agalsidase alfa for 20 months, suggesting patients maintained disease stability. The switch in treatment was generally well tolerated.
These results largely confirm those reported by Tsuboi et al. in a similarly designed study in 11 Japanese patients with Anderson-Fabry disease (Tsuboi and Yamamoto 2012). In their study, eGFR also remained stable when patients switched to agalsidase alfa. Unlike the current study, their echocardiographic findings suggested improvements in LV mass index and wall thickness after 12 months agalsidase alfa treatment compared with agalsidase beta treatment (Tsuboi and Yamamoto 2012). They reported a similar lack of increase in pain symptoms or deterioration in quality of life with the switch in treatment to the current study. In addition, the results of the current study also support those of a retrospective cohort study of the effects of the agalsidase beta shortage on patients with Anderson-Fabry disease, where 20 patients switched to agalsidase alfa, 18 of whom did so only after taking reduced-dose agalsidase beta, and 15 stayed on agalsidase beta but at a reduced dose (Smid et al. 2011). There was no increase in the clinical event incidence rate during the shortage (clinical events included neurological, renal, and cardiac events).
Most, but not all (Koskenvuo et al. 2008; Kovacevic-Preradovic et al. 2008), prior studies that investigated the effects of ERT (agalsidase beta or agalsidase alfa) on cardiac functional parameters reported improvements in cardiac parameters after initiation of ERT (Weidemann et al. 2003; Beck et al. 2004; Hughes et al. 2008; Vedder et al. 2008; Imbriaco et al. 2009). Results of the current study were in agreement with the majority of these studies, i.e., LV mass and wall thickness decreased significantly from pre-ERT values after initiation of ERT, which in this case was with agalsidase beta (retrospective data; see Table 1). In particular, Eng et al. (2001) demonstrated in a placebo-controlled, double-blind study that agalsidase beta (1 mg/kg) resulted in a histological clearance of Gb3 deposits in myocardial endothelial cells; this effect was sustained over 54 months of therapy (Germain et al. 2007). Furthermore, Weidemann et al. suggested that clearance of Gb3 by agalsidase beta leads to a regression of LV hypertrophy, which was documented by echocardiography and confirmed by MRI (Weidemann et al. 2003). Sustained treatment with adequate doses of enzyme is of particular importance as an in vitro study has shown that a relatively limited percentage of administered enzyme reaches the cardiac compartment (Ioannou et al. 2001). The current study did not measure plasma or myocardial Gb3 levels; thus, whether myocardial Gb3 deposits were reduced after sustained ERT for 48 months with agalsidase beta and then 20 months with agalsidase alfa in this study is not known. The Tsuboi et al. switch study did not measure myocardial Gb3 levels either, but their patients experienced an increase in plasma Gb3 levels after the switch from agalsidase beta to alfa treatment (Tsuboi and Yamamoto 2012). Nevertheless, patients still experienced reductions in LV mass after the switch. The authors queried the relevance of Gb3 levels as biomarkers of Fabry disease.
It has been recommended that patients receiving ERT are also prescribed angiotensin converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB) to reduce the risk of proteinuria (Ortiz et al. 2008; Oqvist et al. 2009; Mehta et al. 2010; Warnock et al. 2010). In our study, seven patients were receiving ACEI and/or ARB therapy. Most of these patients started ACEI/ARB therapy at the outset of ERT. Some patients received both ACEI and ARB agents; the dosages of ACEI/ARB therapy and other agents that could affect kidney function and proteinuria were adjusted empirically, depending on individual baseline proteinuria levels, response to antiproteinuric therapy, and tolerance of the antihypertensive effects. Antiproteinuric therapy was continued throughout the study period. We ascribed the significant drop in urinary protein observed during the ERT period to concomitant use of ACEI/ARB therapy and ERT.
The present study has some potential limitations that should be considered, including the relatively small number of evaluated patients and the relatively short time of switching therapy. Further study in larger patient populations is warranted, and according to ClinicalTrials.gov, a switching study with a planned enrolment of 200 patients is under way in the USA (NCT01268241) (ClinicalTrials.gov. 2012). It is the nature of many studies of rare diseases that they are underpowered due to the difficulty in enrolling sufficient eligible patients within a reasonable time frame. However, underpowered studies for rare diseases are justified and ethical in that they allow clinical data to be amassed over time and these data can then be consolidated in meta-analyses, thereby increasing the power and strength of the findings and adding to the clinical evidence base.
Development of neutralizing IgG or IgE antibodies to agalsidase alfa or agalsidase beta were not determined in this study, although switching treatments is not expected to prevent IgG antibody formation or related adverse effects because of complete cross reactivity (Linthorst et al. 2004). Nevertheless, our study is the first MRI study reporting positive long-term effects of switching therapy ERT on cardiac performance in patients with Anderson-Fabry disease.
Conclusions
The results of the present study demonstrate that switching therapy from agalsidase beta to agalsidase alfa, the two products currently available for treatment of patients with Anderson-Fabry disease, did not result in significant changes in renal function or cardiac functional parameters, pain symptoms or health status, and was generally well tolerated. Patients were able to maintain a clinically stable disease state after the switch to long-term agalsidase alfa treatment.
Appendix Details of the Contributions of Individual Authors
AP was primarily involved in patient management, literature search, and preparation of manuscript. BV, IC, MS, and ER assisted in patient management and were involved in manuscript preparation. LS was involved in the literature search. GM performed the cMRI analysis. MI was involved in the literature search and manuscript preparation. All authors read and approved the final manuscript. The results presented in this chapter have not been published previously in whole or part, except in abstract form.
Guarantor
Dr. Antonio Pisani serves as guarantor for the chapter, accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Competing Interest Statement
No authors at any time received payment or services from a third party for any aspect of the submitted work and they have nothing to declare.
Provide Details of Funding
Editorial assistance for manuscript preparation was provided by Tracy Harrison and Mary Hines, in Science Communications, Springer Healthcare.
Details of Ethics Approval and Patient Consent
The study was conducted in accordance with the Declaration of Helsinki and was approved by the institutional ethics committee. All patients switched to agalsidase alfa and included in the final study population provided written informed consent.
Synopsis
Patients with Anderson-Fabry disease can be safely switched from long-term treatment with agalsidase beta to agalsidase alpha and maintain their health status, without any worsening of renal function, cardiac mass, or pain symptoms.
Footnotes
Competing interests: None declared
References
- Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146(2):77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- Beck M, Ricci R, Widmer U, et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest. 2004;34(12):838–844. doi: 10.1111/j.1365-2362.2004.01424.x. [DOI] [PubMed] [Google Scholar]
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- Branton M, Schiffmann R, Kopp JB. Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol. 2002;13(Suppl 2):S139–143. [PubMed] [Google Scholar]
- ClinicalTrials.gov. (2012) The efficacy and safety of switch between agalsidase beta to agalsidase alfa for enzyme replacement in patients with Anderson-Fabry disease (SWITCH). Accessed 3 September 2012, from www.clinicaltrials.gov/ct2/show/NCT01268241
- Desnick R, Ioannou Y, Eng C. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 3733–3774. [Google Scholar]
- Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W. Fabry’s disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J lLab Clin Med. 1973;81(2):157–171. [PubMed] [Google Scholar]
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A–replacement therapy in Fabry’s disease. N Engl J Med. 2001;345(1):9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency (2009) Questions and answers on the shortages of Cerezyme and Fabrazyme. Accessed 1 June 2012, from http://www.emea.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500018393.pdf
- European Medicines Agency (2010) Assessment report for Fabrazyme agalsidase beta. Accessed 1 June 2012, from www.ema.europa.eu/docs/en_GB/document_library/Other/2010/11/wc500099241.pdf
- European Medicines Agency (2010) European Medicines Agency reviews treatment recommendations for Fabrazyme. Accessed 1 June 2012, from http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/10/news_detail_001136.jsp&mid=WC0b01ac058004d5c1
- Feriozzi S, Torras J, Cybulla M, et al. The effectiveness of long-term agalsidase alfa therapy in the treatment of Fabry nephropathy. Clin J Am Soc Nephrol. 2012;7(1):60–69. doi: 10.2215/CJN.03130411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18(5):1547–1557. doi: 10.1681/ASN.2006080816. [DOI] [PubMed] [Google Scholar]
- Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008;94(2):153–158. doi: 10.1136/hrt.2006.104026. [DOI] [PubMed] [Google Scholar]
- Imbriaco M, Pisani A, Spinelli L, et al. Effects of enzyme-replacement therapy in patients with Anderson-Fabry disease: a prospective long-term cardiac magnetic resonance imaging study. Heart. 2009;95(13):1103–1107. doi: 10.1136/hrt.2008.162800. [DOI] [PubMed] [Google Scholar]
- Ioannou YA, Zeidner KM, Gordon RE, Desnick RJ. Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. Am J Hum Genet. 2001;68(1):14–25. doi: 10.1086/316953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann C, Linhart A, Baehner F, et al. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130(3):367–373. doi: 10.1016/j.ijcard.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Koskenvuo JW, Hartiala JJ, Nuutila P, et al. Twenty-four-month alpha-galactosidase A replacement therapy in Fabry disease has only minimal effects on symptoms and cardiovascular parameters. J Inherit Metab Dis. 2008;31(3):432–441. doi: 10.1007/s10545-008-0848-3. [DOI] [PubMed] [Google Scholar]
- Kovacevic-Preradovic T, Zuber M, Attenhofer Jost CH, et al. Anderson-Fabry disease: long-term echocardiographic follow-up under enzyme replacement therapy. Eur J Echocardiogr. 2008;9(6):729–735. doi: 10.1093/ejechocard/jen129. [DOI] [PubMed] [Google Scholar]
- Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- Lidove O, West ML, Pintos-Morell G, et al. Effects of enzyme replacement therapy in Fabry disease–a comprehensive review of the medical literature. Genet Med. 2010;12(11):668–679. doi: 10.1097/GIM.0b013e3181f13b75. [DOI] [PubMed] [Google Scholar]
- Linhart A. Treatment of Anderson-Fabry disease. Heart. 2008;94(2):138–139. doi: 10.1136/hrt.2006.113886. [DOI] [PubMed] [Google Scholar]
- Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007;93(4):528–535. doi: 10.1136/hrt.2005.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linthorst GE, Germain DP, Hollak CE, et al. Expert opinion on temporary treatment recommendations for Fabry disease during the shortage of enzyme replacement therapy (ERT) Mol Genet Metab. 2011;102(1):99–102. doi: 10.1016/j.ymgme.2010.11.155. [DOI] [PubMed] [Google Scholar]
- Linthorst GE, Hollak CEM, Donker-Koopman WE, Strijland A, Aerts JMFG. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66(4):1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- Mehta A, Beck M, Elliott P, et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374(9706):1986–1996. doi: 10.1016/S0140-6736(09)61493-8. [DOI] [PubMed] [Google Scholar]
- Mehta A, West ML, Pintos-Morell G, et al. Therapeutic goals in the treatment of Fabry disease. Genet Med. 2010;12(11):713–720. doi: 10.1097/GIM.0b013e3181f6e676. [DOI] [PubMed] [Google Scholar]
- Melzack R. The short-form McGill pain questionnaire. Pain. 1987;30(2):191–197. doi: 10.1016/0304-3959(87)91074-8. [DOI] [PubMed] [Google Scholar]
- Morrissey RP, Philip KJ, Schwarz ER. Cardiac abnormalities in Anderson-Fabry disease and Fabry’s cardiomyopathy. Cardiovascular J Africa. 2011;22(1):38–44. [PMC free article] [PubMed] [Google Scholar]
- Oqvist B, Brenner BM, Oliveira JP, et al. Nephropathy in Fabry disease: the importance of early diagnosis and testing in high-risk populations. Nephrol, Dialysis, Transplant: Off Publ Eur Dialysis Transplant Assoc - Eur Renal Assoc. 2009;24(6):1736–1743. doi: 10.1093/ndt/gfp105. [DOI] [PubMed] [Google Scholar]
- Ortiz A, Oliveira JP, Wanner C, Brenner BM, Waldek S, Warnock DG. Recommendations and guidelines for the diagnosis and treatment of Fabry nephropathy in adults. Nat Clin Pract Nephrol. 2008;4(6):327–336. doi: 10.1038/ncpneph0806. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Kopp JB, Austin HA, 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- Smid BE, Rombach SM, Aerts JMFG, et al. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients. Orphanet J Rare Dis. 2011;6:69. doi: 10.1186/1750-1172-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Yamamoto H (2012) Clinical observation of patients with Fabry disease after switching from agalsidase beta (Fabrazyme) to agalsidase alfa (Replagel). Genetics Med [Epub ahead of print:doi:101038/gim/2012/39] [DOI] [PMC free article] [PubMed]
- Vedder AC, Breunig F, Donker-Koopman WE, et al. Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab. 2008;94(3):319–325. doi: 10.1016/j.ymgme.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Ware JE. SF-36 health survey: manual and interpretation guide. Boston, MA: The Health Institute, New England Medical Centre; 1997. [Google Scholar]
- Warnock DG, Daina E, Remuzzi G, West M. Enzyme replacement therapy and Fabry nephropathy. Clin J Am Soc Nephrol. 2010;5(2):371–378. doi: 10.2215/CJN.06900909. [DOI] [PubMed] [Google Scholar]
- Weidemann F, Breunig F, Beer M, et al. The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. Eur Heart J. 2005;26(12):1221–1227. doi: 10.1093/eurheartj/ehi143. [DOI] [PubMed] [Google Scholar]
- Weidemann F, Breunig F, Beer M, et al. Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation. 2003;108(11):1299–1301. doi: 10.1161/01.CIR.0000091253.71282.04. [DOI] [PubMed] [Google Scholar]
- Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75(1):65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate YA, Hopkin RJ. Fabry's disease. Lancet. 2008;372(9647):1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]