

Abstract
A 5 years old boy affected with Glycogen Storage Disease type Ia (GSD-Ia) with previous optimal metabolic control developed severe erratic hypoglycemic episodes during continuous nocturnal gastric drip-feeding (CNGDF) administered by nasogastric tube. The episodes of hypoglycemia were not related to pump failure or human errors or wrong position of the tube in the gastrointestinal tract. Hyperinsulinism was also considered in this patient but it was excluded mainly because hypoglycemia was only nocturnal. Moreover, hypoglycemic episodes disappeared when CNGDF was stopped and he was fed with normal meals. The fact that hypoglycemia resolved after stopping CNGDF when nocturnal meals were introduced led us to hypothesize that CNGDF rich with simple carbohydrates might have been the cause of a sort of dumping-like syndrome. Dumping syndrome (DS) develops when a large amount of carbohydrate reaches the small intestine due to rapid gastric emptying (Tack et al. 2009; Hejazi et al. 2010). We suppose that CNGDF induced a disturbance of gastric motility with a gastric accumulation of fluids at a certain time of the night followed by a rapid voiding of the stomach leading to DS.
Introduction
Glycogen Storage Diseases (GSDs) are a group of inherited disorders characterized by enzyme defects that affect the glycogen metabolism. The deficiency of the glucose-6 phosphatase (EC 3.1.3.9) activity causes Glycogen Storage Disease type Ia (GSD-Ia, MIM 232200) that produces fasting hypoglycemia, hyperlacticacidemia, hyperlipidemia and hyperuricemia, growth retardation, enlargement of kidneys and progressive renal involvement, and risk of development of hepatocellular adenoma and carcinoma.
The current treatment of GSD-Ia is frequent diurnal small meals followed by orally administered uncooked cornstarch, a complex carbohydrate composed of highly branched glucose chains and slowly hydrolyzed in the small intestine, and continuous nocturnal gastric drip-feeding (CNGDF) through nasogastric tube or through gastrostomy with maltodextrins alone or free-lactose milk and maltodextrins. Diet composition is recommended to be hypolipidic, with normal protein and elevated carbohydrates intake. Carbohydrates are all complex during the day and maltodextrins are given continuously through the night. These measures are effective in maintaining the metabolic control (Rake et al. 2002) although growth retardation and liver, renal, and other complications may still occur in the long-term follow-up even in well-controlled patients (Chen 2001; Heller et al. 2008; Melis et al. 2010).
Suggested rates of CNGDF are different at various ages on the basis of different energy needs: a rate of 7–9 mg/kg/min of carbohydrates is recommended in infants below 1 year of age, 6–8 mg/kg/min in children younger than 6 years, 5–6 mg/kg/min in school children and adolescents, and 3–4 mg/kg/min in adults (Rake et al. 2002).
Dumping syndrome (DS) consists of early (abdominal and vasomotor) and late (hypoglycemia) symptoms (Tack et al. 2009) and results from rapid delivery of simple carbohydrate-rich osmotic fluids to the small intestine. In normal conditions, the presence of glucose in the jejunum is a powerful stimulus for insulin secretion. Hyperosmolar significant amount of glucose in the duodenum substantially produces two effects: one is shift of fluids from the intravascular component to the duodenum leading to release of vasoactive agents and probably responsible of early symptoms of DS, such as tachycardia, nausea, abdominal pain, and hypotension; the other is a hyperinsulinemic response inducing hypoglycemia, the late effect of DS (Tack et al. 2009; Hejazi et al. 2010).
DS has mainly been described in children after surgical procedures for gastroesophageal reflux, like Nissen fundoplication, or in rare cases of microgastria, partial or total gastrectomy, accidental intraduodenal or jejunal administration of bolus feeding, or inadequate meals with high osmolarity, as well as rare cases of generalized autonomic dysfunction in two children operated for esophageal atresia without gastroesophageal reflux (Michaud et al. 2010), these last indicating that only esophageal dysmotility may be the trigger point. DS is also diagnosed in patients with type 2diabetes mellitus and in many patients previously diagnosed as cyclic vomiting syndrome (Hejazi et al. 2010).
We here report the case of a patient with GSD-Ia who had several unexplained episodes of hypoglycemia during CNGDF administered by nasogastric tube. Hypoglycemic episodes disappeared when CNGDF was stopped and he was fed with normal meals. By exclusion of other causes of hypogycemia, we hypothesize that this patient had a form of dumping-like syndrome caused by a gastric motility disorder associated to CNGDF.
Case Report
The patient is the first male child of nonconsanguineous Italian parents. He was born via a spontaneous vaginal delivery at 39 weeks gestational age with birth weight of 3.150 kg. His perinatal period was normal. At 4 months of life, he was hospitalized for frequent regurgitation, failure to thrive, and severe hepatomegaly (at the umbilical plane). On the basis of the clinical picture, the finding of fasting hypoglycemia, hypertriglyceridemia, and hypercholesterolemia and hyperechogenic liver tissue at ultrasound, a GSDs was suspected and later GSD-Ia was confirmed through the molecular analysis of glucose-6 phosphatase gene (G6PC) which showed that the patient carried the mutations p.Y172X/p.W86C.
The boy was regularly followed and maintained optimal growth and metabolic control of the disease with frequent meals, followed by cornstarch and CNGDF. At the age of 4 years and 6 months, he had mild hepatomegaly (1 cm from rib cage) and normal psychomotor development, the weight was 16.0 kg (25th centile) and the height was 103 cm (25th–50th centile).
His diet consisted of five diurnal meals every 3–4 h with cornstarch 35–40 g/day and CNGDF prepared with 15 g of lactose-free milk AL 110 (Nestlè, Milan, Italy), 70 g of maltodextrins powder Fantomalt (Nutricia, Milan, Italy) in 420 mL of water at rate 42 mL/h (8.31 mg/kg/min). Intake of glucose/kg/min with CNGDF (8–10 mg/kg/min) was elevated for patient’s age because he has always been a lean, very active child with augmented energy needs.
He had normal pre-prandial glucose levels (70–90 mg/dL), no post-prandial hyperglycemia (90–100 mg/dL), normallactate (2.6-3.3 mmol/L, normal range 0.44-2.22 mmol/L), triglycerides (71–159 mg/dL, normal range 50–200 mg/dL), cholesterol (93–136 mg/dL, normal range 130–200 mg/dL), and uric acid (3.8-5.2 mg/dL, normal range 3–7 mg/dL).
At the age of 5 years, he had a generalized seizure with hypoglycemia (23 mg/dL measured with finger-stick glucose at home) at 4 am while he was fed with CNGDF; he had a rapid resolution after the administration of 30 mL of 33 % glucose solution via nasogastric tube. The child was then admitted to the local hospital. Feeding-pump functioning was verified and the nasogastric tube was repositioned. Rate of CNGDF was temporarily increased to 110 mL/h and then modified to maintain blood glucose in the normal range but he had another generalized seizure at 8.30 am caused by hypoglycemia (34 mg/dL) during the CNGDF (rate 45 mL/h).
After the resolution of the symptoms and the improvement of blood glucose, the patient was discharged that day and the parents were instructed in measuring glycemia with finger-stick glucose monitoring every hour during the night. The night after, the patient had another asymptomatic hypoglycemia at home (39 mg/dL at 4 am) during CNGDF (rate 50 mL/h). He was then accompanied to our Department. In his first night in our hospital, he had an important asymptomatic hypoglycemia (26 mg/dL at 3.30 am) during CNGDF prepared with 34 g of lactose-free milk AL 110, 96 g of maltodextrins powder Polycose in 600 mL of water at a rate of 55 mL/h (10.8 mg/kg/min). The day after, he had another asymptomatic hypoglycemia (39 mg/dL) at 4.15 am and in this occasion growth hormone (0.11 ng/mL, normal range 0–10 ng/mL), insulin (1.0 μUI/mL, normalrange 2.6–24.9 μUI/mL), C-peptid (1.1 ng/mL, normal range 0.8–4.2 ng/mL), and cortisol (99.6 ng/mL, normal range 62–194 ng/mL) were analyzed and resulted normal. We checked with X-rays that the tube was correctly positioned in the stomach. Monitoring of blood glucose and insulin during the day, immediately pre-prandial, and 1 h later did show an optimal metabolic profile for a GSD-Ia patient (Fig. 1). Munchausen syndrome was excluded first for the fact that different relatives were staying in hospital with the child (father, mother and grandmother) in the nights when he had hypoglycemias and furthermore because the psychologist excluded this possibility and defined this family as very well structured and optimally self- supporting.
At this point the composition and the rate of CNGDF were changed (35 g of lactose-free milk AL 110, 100 g of maltodextrins powder Polycose in 625 mL of water at 40 mL/h from 9.30 pm to 00.00 and at increased rate to 60 mL/h from 00.00 to 7.30 am): an improvement of nocturnal blood glucose was observed for 4 days and the patient was discharged.
At home, 18 days after the discharge, he experienced three other hypoglycemias during CNGDF (rate 55–60 mL/h), one with generalized seizure (35 mg/dL) and the other two asymptomatic (39 mg/dL, 46 mg/dL). The episodes of hypoglycemia in hospital and at home monitored with finger-stick glucose are reported in Fig. 2. Figure 3a shows results of continuous glucose monitoring (Guardian REAL-Time CGM System, Medtronic, Northridge, USA) at home during CNGDF. In these nights no hypoglycemia was detected but blood glucose values were fluctuating much more than we would expect with a CNGDF.
Fig. 1.

Monitoring of blood glucose (solid line) and insulin (broken line) during the day, immediately pre-prandial and 1 h later (9 meals). We observed optimal values for a GSD-Ia patient metabolic profile
Fig. 2.

Nocturnal blood glucose concentration with finger-stick glucose monitoring (●) and the rate of CNGDF (■). We report five episodes of nocturnal hypoglycemias during 4 nights of CNGDF
Fig. 3.
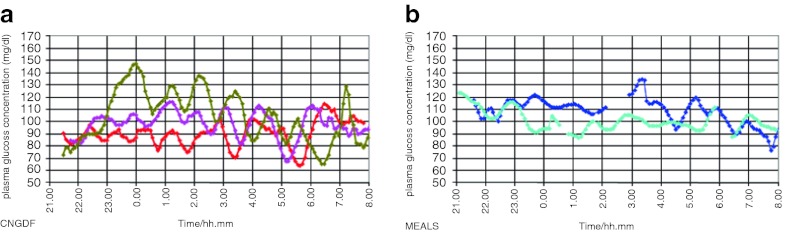
Continuous monitoring of nocturnal blood glucose with Glycemic Holter: we report in Fig. 3a blood glucose of 3 nights during CNGDF and in Fig. 3b blood glucose of 2 nights during nocturnal meals. During CNGDF, glucose values in this patient show important variations with significant up and down even when he does not reach hypoglycemic values. Glucose blood variations are less relevant with nocturnal meals
Then we stopped CNGDF and introduced nocturnal meals with 7 g of lactose-free milk AL 110, 8 g of multicereal cream and 40 g of uncooked cornstarch maizena at 10.30 pm and 3.30 am and 15 g of lactose-free AL110 and 25 g of uncooked cornstarch maizena at 7.30 am. The patient did not have any other nocturnal hypoglycemia. The continuous glucose monitoring with meals at home is reported in Fig. 3b. Surprisingly it was less fluctuating than the profile obtained with CNGDF.
Nowadays, at the age of 12 years, the boy is still having frequent meals during the night and has an optimal metabolic balance (last triglycerides 78 mg/dL, cholesterol 163 mg/dL, uric acid 3.6 mg/dL and lactate 2.1 mmol/L) and his dietary treatment consists of seven meals at 7.15 am, 10.45 am, 1.15 pm, 5.15 pm, 7.00 pm, 10.30 pm, and 3.15 am with a glucose intake of 7 mg/kg/min.
From the age of 6 years and 6 months, the patient presented glomerular hyperfiltration without microalbuminuria and proteinuria and we started at the age of 10 years the treatment with Ramipril 1.25 mg/day, incremented to 2.5 mg/day for persistent hyperfiltration without hypotension.
Discussion
Our patient had severe repeated episodes of symptomatic hypoglycemia while he was fed through CNGDF prepared with a high amount of simple carbohydrates. The rationale of giving simple carbohydrates continuously during the night relies on the assumption that CNGDF is associated with regular continuous absorption of nutrients.
We first excluded human errors, misfunctioning of the feeding pump, tube entanglement or blockages, faulty equipment, and child tampering with pumps and feeding equipment. Our second hypothesis was that of a wrong position of the tube in the gastrointestinal tract but, as the tube was replaced every night and taken away in the morning, we should assume that every night, at a certain time, the tip of the tube went down to the duodenum and this is not easily happening. When we checked with X-rays, the tube was correctly positioned in the stomach.
Hyperinsulinism was also considered: in fact sometimes GSD-Ia patients who are excessively fed can manifest hypoglycemia around 2–2.5 h after the meal but it does not happen during CNGDF. However, this child was never hypoglycemic during the day but only during CNGDF. Even though he had quite an elevated intake of glucose/kg/min for his age, he was really thin and much physically active every day; he was also optimally controlled from the metabolic point of view, with normal blood lactate, triglycerides, and uric acid. For all these reasons we had to exclude that this patient was affected by hyperinsulinism. We also excluded a Munchausen syndrome: the patient was followed by different relatives when had hypoglycemias in the hospital and the family was well structured from a psychological point of view.
We first tried to modify composition and rate of delivery of CNGDF in this child without success. Only after all these unsuccessful trials, considering that hypoglycemia occurred only during CNGDF, we decided ex juvantibus to stop the CNGDF and substitute it with meals. The fact that hypoglycemia resolved after the introduction of nocturnal meals led us to suppose that CNGDF rich with simple carbohydrates might have been the cause of a sort of dumping-like syndrome. Our interpretation is that it may be possible that this kind of artificial feeding during the night interfered with gastric motility and induced at a certain time of the night an accumulation of fluids, followed by a rapid voiding of the stomach leading to DS.
DS is a constellation of gastrointestinal and systemic symptoms secondary to volume shifts and excessive release of insulin and various vasoactive peptides due to abnormal emptying of gastric content (Tack et al. 2009).
The physiopathology of DS is still controversial and probably multifactorial. The rapid passage of gastric content, especially if it is rich in small molecules, into the small bowel results in a shift of fluids and electrolytes into the intestinal lumen, loss of intravascular volume, and small bowel distension leading to the release of gastrointestinal hormones (Caulfield et al. 1987; Samuk et al. 1996). The rush of fluid can cause bowel stretching and cramps, nausea and vomiting, bloating, and diarrhea (early dumping syndrome). Late DS occurs 1–2 h after the meal and is characterized by systemic vascular symptoms including flushing, dizziness, weakness, sweating, fainting and palpitation, fall in blood pressure, and increase of heart rate (Hejazi et al. 2010). The rapid absorption of glucose is countered by excessive release of insulin responsible for the subsequent reactive hypoglycemia (Hejazi et al. 2010).
DS is suspected according to symptoms and medical history and confirmed through measurement of gastric emptying by scintigraphy. A 4 h Gastric Emptying Test (GET) using a low fat meal is the gold standard to investigate DS (Hejazi et al. 2010). However, this diagnostic method is rarely used in pediatric population and we lacked normal range values for this age. Second, this patient had symptoms only during the CNGDF and it was clear that he did not have problems after a normal meal. Third, the standard meal that is suggested for this test is not appropriate to a GSD-Ia patient. Another test which is suggested for DS diagnosis is dumping provocation by the oral glucose challenge (50 g after 10-h fasting): higher plasma levels of glucose during the first 60 min after provocation and reduced plasma glucose levels 60–180 min later have been used as diagnostic criteria for DS (Ukleja 2005).
The treatment of DS primarily consists of dietary manipulations as limiting fluid intake while eating solid small meals, high in protein and fat and low in carbohydrates, increasing meal viscosity and assuming a recumbent position following a meal (Bouras and Scolapio 2004). Pectins, glucomannan, and guar gum have been tested with good results, especially in the pediatric population (Tack et al. 2009). Uncooked cornstarch after the meals has been shown to be effective in a small number of children (Gitzelmann and Hirsig 1986; Borovoy et al. 1998).
We decided not to perform diagnostic conclusive tests in this child because they were too demanding for him and his family and because we had already obtained the resolution of symptoms. DS has mainly been described in children after surgical procedures on the gastrointestinal tract, and in few rare cases of generalized autonomic dysfunction in two children operated for esophageal atresia without gastroesophageal reflux (Michaud et al. 2010). In analogy with these few cases we hypothesized a posteriori that CNGDF could have been the cause of gastro-esophageal dysmotility in this child. It appeared to be confirmed by the trace of continuous nocturnal glucose monitoring which showed more relevant ups and downs during CNGDF than when the patient was fed with nocturnal meals (Fig. 3). This finding is enough to say that, despite continuous feeding with simple carbohydrates, the absorption of glucose during the night was not continuous. The explanation might only be a disturbance of gastrointestinal motility.
In conclusion, we present a 5-year-old GSD-Ia patient with previously optimal metabolic control who developed severe erratic hypoglycemic episodes during CNGDF. By exclusion, we hypothesized a disturbance of gastric motility during CNGDF.
We think that the more reasonable explanation of these findings is that nasogastric drip feeding induced a disturbance of gastric motility. It would be interesting to know if other cases exist with these symptoms and whether they are related to the use of nasogastric tube instead of gastrostomy.
Acknowledgments
We gratefully acknowledge the family and the child, the nurses of the Pediatric Dept in San Gerardo Hospital, and the secretary of the Rare Metabolic Diseases Unit, Vera Marchetti.
Abbreviations
- CNGDF
Continuous nocturnal gastric drip-feeding
- DS
Dumping syndrome
- G6PC
Glucose-6 phosphatase gene.
- GET
Gastric emptying test
- GSD-Ia
Glycogen storage disease type Ia
- GSDs
Glycogen storage diseases
Footnotes
Competing interests: None declared
Contributor Information
A. Brambilla, Email: alebramby@libero.it
A. Pozzoli, Email: a.pozzoli@ausl.pc.it
F. Furlan, Email: f.furlan@hsgerardo.org
R. Parini, Email: rossella.parini@unimib.it
References
- Borovoy J, Furuta L, Nurko S. Benefit of uncooked cornstarch in the management of children with dumping syndrome fed exclusively by gastrostomy. Am J Gastroenterol. 1998;93(5):814. doi: 10.1111/j.1572-0241.1998.231_a.x. [DOI] [PubMed] [Google Scholar]
- Bouras EP, Scolapio JS. Gastric motility disorders. Management that optimizes nutritional status. J Clin Gastroenterol. 2004;38:549–557. doi: 10.1097/00004836-200408000-00003. [DOI] [PubMed] [Google Scholar]
- Caulfield ME, Wyllie R, Firor HV, Michener W. Dumping syndrome in children. J Pediatr. 1987;110:212–215. doi: 10.1016/S0022-3476(87)80156-7. [DOI] [PubMed] [Google Scholar]
- Chen YT, et al. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular basis of inherited diseases. New York: McGraw-Hill; 2001. pp. 1521–1552. [Google Scholar]
- Gitzelmann R, Hirsig J. Infant dumping syndrome: reversal of symptoms by feeding uncooked starch. Eur J Pediatr. 1986;145:504–506. doi: 10.1007/BF02429052. [DOI] [PubMed] [Google Scholar]
- Hejazi RA, Patil H, McCallum RW. Dumping syndrome: establishing criteria for diagnosis and identifying new etiologies. Dig Dis Sci. 2010;55:117–123. doi: 10.1007/s10620-009-0939-5. [DOI] [PubMed] [Google Scholar]
- Heller S, Worona L, Consuelo A. Nutritional therapy for glycogen storage diseases. JPGN. 2008;47:S15–S21. doi: 10.1097/MPG.0b013e3181818ea5. [DOI] [PubMed] [Google Scholar]
- Melis D, Pivonello R, Parenti G, et al. The growth hormone-insulin-like growth factor axis in glycogen storage disease type 1: evidence of different growth patterns and insulin-like growth factor levels in patients with glycogen storage disease type 1a and 1b. J Pediatr. 2010;156(4):663–670. doi: 10.1016/j.jpeds.2009.10.032. [DOI] [PubMed] [Google Scholar]
- Michaud L, Sfeir R, Couttenier F, Turck D, Gottrand F. Dumping syndrome after esophageal atresia repair without antireflux surgery. J Pediatr Surg. 2010;45:E13–E15. doi: 10.1016/j.jpedsurg.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GPA. Guidelines for management of glycogen storage disease type I. European study on glycogen storage disease type I (ESGSD I) Eur J Pediatr. 2002;161:S112–S119. doi: 10.1007/BF02680007. [DOI] [PubMed] [Google Scholar]
- Samuk I, Afriat R, Horne T, Bistritzer T, Barr J, Vinograd I. Dumping syndrome following Nissen fundoplication, diagnosis and treatment. J Pediatr Gastroenterol Nutr. 1996;23:235–240. doi: 10.1097/00005176-199610000-00006. [DOI] [PubMed] [Google Scholar]
- Tack J, Arts J, Caenepeel P, De Wulf D, Bisschops R. Pathophysiology, diagnosis and management of postoperative dumping syndrome. Nat Rev Gastroenterol Hepatol. 2009;6(10):583–590. doi: 10.1038/nrgastro.2009.148. [DOI] [PubMed] [Google Scholar]
- Ukleja A. Dumping syndrome: pathophysiology and treatment. Nutr Clin Pract. 2005;20(5):517–525. doi: 10.1177/0115426505020005517. [DOI] [PubMed] [Google Scholar]