

Abstract
Sarcosinemia is a rare inborn error of metabolism that is characterised by an increased level of sarcosine (N-methylglycine) in the plasma and urine. The enzymatic block results from a deficiency of sarcosine dehydrogenase (SarDH), a liver mitochondrial matrix enzyme that converts sarcosine into glycine. Although this condition may remain inapparent until later life, it has been reported in rare cases to lead to neurodevelopmental disability. A 19-year-old male with sarcosinemia presented with dystonia, developmental delay and cognitive impairment. Magnetic resonance imaging revealed vermian hypotrophy. A 2-year pharmacological treatment with memantine was negative on the clinical signs. In this case, it was concluded that the metabolic block leading to sarcosinemia was responsible of a pathologic condition with mental deficiency and complex neurological signs. A maternal isodisomy discovered in the vicinity of SarDH gene could contribute to this pathology. Deficit of SarDH may be considered as a differential diagnosis of growth failure during prenatal stages and respiratory failure at birth following a slowly progressive developmental delay.
Introduction
Sarcosinemia (OMIM 268900) is a rare autosomal recessive inborn error of the one-carbon metabolism, with increased plasma level of sarcosine and high urine excretion. Sarcosine (N-methylglycine) is synthesised from dimethylglycine by dimethylglycine dehydrogenase and converted to glycine by sarcosine dehydrogenase (SarDH), within the mitochondrial matrix. Since the first case published in 1966 by Gerrsitsen and Waisman (1966), over around 30 patients were reported. The presentation is mostly by mental delay, but also cardiomyopathy, deafness and visual disturbance. Occasionally, deficit of SarDH might remain asymptomatic until later life (Christensen et al. 1989).
We report the case of 19 year old male in whom a sarcosinemia was diagnosed on a complex neurological syndrome developing progressively since childhood with a history of developmental delay. A follow-up of 18 months pharmacological trial is discussed.
Report of a Case
A 19 year young male was referred to our clinic because of the worsening of his neurological condition. From the parents, we learned that he had a long and progressive history of neurological impairment. Our patient was delivered prematurely at 34 weeks of uncomplicated gestation to non-consanguineous parents. The infant was born small for date (weight 1,200 g, occipitofrontal circumference 28 cm, length 43 cm; –2 σ) but morphologically normal. He required a short ventilatory support because of mild respiratory difficulties and lethargy at birth. Because of low growth and reduced sucking he stayed in a continuous care unit during the first 2 months. Slight intermittent dystonic posture of the right leg and muscular hypotonia were noted in the first 14 months of his life. At the age of two, he started to walk but did not speak. Since the first school classes his education results were considered as weak but he learned to write. He was considered as awkward, with motor and speech disabilities (intelligence quotient of 56 in verbal and 57 in performance). Due to these difficulties, our patient had to attend a specialised school. At about the age of 9, gait progressively worsened due to dystonia and postural instability. He showed emotional lability with rare violent outbursts and behavioural problems. Around the age of 16, he progressively developed a dysarthria with blurred and slow speech and cerebellar ataxia. Otherwise, his neurological condition remained stable, without any acute exacerbation. At the age of 19, gait disturbance and dystonic movements became permanent, leading to hospitalisation. The physical parameters remained harmoniously low: occipitofrontal circumference of 53 cm (–2 σ), weight 43 kg (–3 σ) and height 163 cm (–1.7 σ). Other neurological symptoms included hyperreflexia without Babinski sign, hypotonia and dystonia exaggerated by stress and present both at rest and walking. There was a hollow foot with permanent internal rotation of the right leg. The dystonic posture of the body and the arms interfered with volitional movements and resulted in a clumsy gait, dropping and spilling, without losing ability to ambulate. Speech was slow, lacking normal prosody. Ophthalmological investigation revealed iris atrophy, congenital left eye cataract and poor saccadic adaptation.
Until this late period, he was considered as having late-onset atypical complications of a perinatal hypoxia, because of the history of respiratory distress at birth followed by complex neurological signs. However, since he was hypotrophic at birth, we considered that a developmental or metabolic illness should be considered. As such, we performed detailed laboratory and brain imaging analysis.
The MRI showed mild vermian atrophia without significant abnormality in the brainstem and the cerebrum or abnormalities in the myelination (Fig. 1a, b). The spectral profile within the pallidum was normal on a 1H magnetic resonance spectrometry (1H-MRS) (Fig. 1c, d).
Fig. 1.
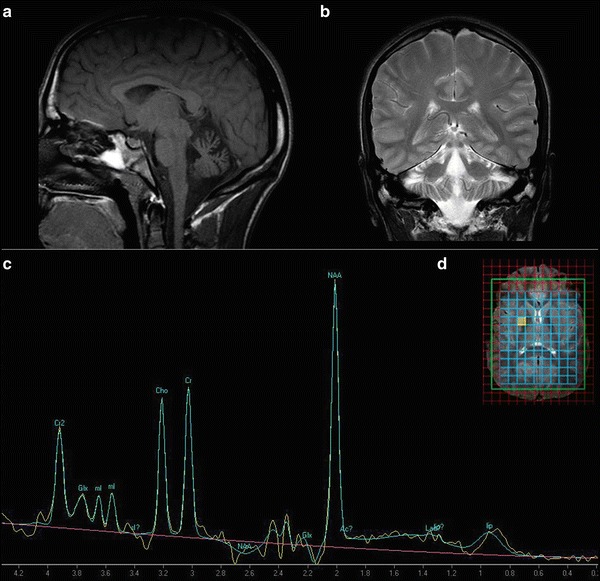
A moderate antero-superior vermian atrophy on sagittal T1-weighted (TR/TE 425 ms/10 ms) (a) and on a coronal section on T2-weighted (b) brain MRI. The scan was performed before the memantine treatment. (c) Proton MR spectroscopy (1H-MRS) at the age of 21 (clinical system of 3T, TE: 30 ms, using commercially available software) reveals no significant changes at short TE. (d) Representative location of the MRS voxels on axial T2-weighted image within the basal ganglia
Basic neuropsychological evaluation performed using standard test battery for intelligence, attention, visuo-spatial and executive functions showed a moderate dysexecutive syndrome with slow information processing speed, impairment of attention and planification processes. A 4 year follow-up showed a stable performance on global intelligence quotient assessment.
Ancillary blood tests including folic acid were within the reference intervals. A chromatography of urine showed an increased excretion of sarcosine at 1,214 μmol/mmol of creatinine. Excessive levels of sarcosine were found in plasma (319 μmol/L) and in CSF (7 μmol/L). All other amino acids were present at normal levels both in the plasma and urine except a slightly higher excretion of glycine considered as dependent of dietary factors (Table 1). Studies for inborn metabolic errors including organic acids, purines and pyrimidines, lactic and pyruvic acid were also normal in plasma, urine and CSF, thus excluding a possible glutaric aciduria type II. Thus, we raised the biochemical diagnosis of possible sarcosinemia. We found no abnormal blood and urine sarcosine levels in the other two siblings, the mother or the father.
Table 1.
Results of biochemical analysis. Nd: not determined.
| Amino acids | Dec./ 07 | Feb./ 08 | Sept./08 | Sept./09 |
|---|---|---|---|---|
| Sarcosine (plasma) μmol/L (reference values <10) | 319 | 326 | 379 | 238 |
| Glycine (plasma) μmol/L (reference values =120–320) | 254 | 255 | 291 | 310 |
| Serine (plasma) μmol/L (reference values = 50–200) | 145 | 135 | 116 | 133 |
| Sarcosine (urine) μmol/mmol of creatinine (reference values < 50) |
1214 | 655 | 631 | nd |
| Glycine (urine) μmol/mmol of creatinine (reference values = 60–190) |
419 | 236 | 198 | nd |
| Serine (urine) μmol/mmol of creatinine (reference values = 20–50) |
77 | 52 | 38 | nd |
| Sarcosine (CSF) μmol/L (reference values = undetectable) |
nd | 7 | 12 | 12 |
| Glycine (CSF) μmol/L (reference values = 7–11) | nd | 6 | 7 | 7 |
| Serine (CSF) μmol/L (reference values = 24–44) | 32 | 28 | 29 | |
| Treatment with memantine | no | no | M7 | M19 |
Since sarcosine is a potent competitive inhibitor of glycine type 1 transporter (GlyT1) (Smith et al. 1992 May), we hypothesised that neurological and cognitive impairment in sarcosinemia could be related to an interference with NMDA receptors. Thus an NMDA receptor antagonist such as memantine could be considered as a symptomatic treatment option. With the informed consent of his parents, a memantine was given for 2 years (20 mg/day the first year, then 40 mg/day), in addition to a rehabilitation programme and orthopaedic treatment on the right leg. Loading tests with folic acid or riboflavin were not performed because of weak therapeutic efficiency reported (van Sprang et al. 1986).
No significant improvement of gait or cognitive performances was noted during the period of treatment with memantine. However, amino acid chromatography analysis of plasma, urine and CSF during the trial revealed some slight lowering of plasma levels and elevation in the CSF (Table 1).
Comment
We present a patient with movement disorder and developmental disabilities suggestive of brain damage where the biochemical analysis revealed sarcosinemia. The incidence of sarcosinemia is difficult to establish because as few as 30 cases were published during the last 50 years. No brain imaging data or follow-up information is available for the previously published cases. Since so few cases have been reported with sparse clinical descriptions, a detailed clinical phenotype could not be extracted from the literature. The only common feature was a low cognitive global performance. Although in some cases sarcosinemia was considered as a benign condition, usually a neurodevelopmental disability starts insidiously, before the brain is fully mature, and progresses slowly over several years. Our patient shares some symptoms (premature birth with respiratory distress, short stature and mental retardation) with previously reported cases of sarcosinemia. The neuropsychological profile is not specific and is similar to the “cerebellar cognitive affective syndrome” (Schmahmann and Sherman 1998; Botez-Marquard et al. 1994). MRI revealed atrophy within the vermis and the anterior cerebellum. The lack of more obvious abnormalities in other parts of the brain suggests that myelination, dendritic arborisation and synaptogenesis are not impaired.
Other diagnostic options such as post-hypoxic encephalopathy or atypical neurodegeneration with brain iron accumulation (NBIA) were discussed. The mild respiratory difficulties at birth could hardly be responsible for significant hypoxic brain damage. None of the morphological or demyelinating sequelae usually reported in hypoxic brain damage (Rees and Inder 2005) was detected on repetitive brain imaging. In addition, we found no report of isolated cerebellar atrophy with slowly progressive late-onset ataxia occurring after brain anoxia in premature infants (Rees and Inder 2005). Furthermore, the low morphometric parameters at birth suggest that the clinical events were conditioned intra uteri, before the eventual perinatal hypoxia.
NBIA is a progressive extrapyramidal neurodegeneration, responsible for at least three different clinical forms always associated with abnormalities on MRI suggesting iron accumulation (Gregory et al. 2009). Considering the MRI images, an atypical NBIA due to PLA2G6 mutation was suggested (A. Gregory, personal communication 2011). Further genetic studies on PLA2G6 were not proposed with a view to the insignificant signal differences on the brain imaging in our patient compared to age-matched controls and the recent genetic data in our patient (Bar-Joseph et al. 2012).
The glycine levels in plasma and CSF in our patient were within the normal ranges except in the initial assay (Table 1). Still, we are not certain whether this assay is the appropriate measure of the potential pathological mechanism of sarcosinemia.
So far, the pathogenesis of sarcosinemia remains unexplained. The SarDH was initially identified in human brain but has high hepatic expression. The SARDH gene locus is on 9q34, which contains 21 exons and spans about 75 kb. A recent paper reports that our patient carries two maternal alleles in the vicinity of the 9q34 (Bar-Joseph et al. 2012). Even though no mutations were detected in the SARDH gene, this maternal isodisomy could be responsible for the sarcosinemia. These data may be helpful for early diagnosis and adequate nutrition early in life.
The clinical manifestations of our patient developed in a continuum of gradually impaired CNS dysfunctions of the structures involved in the movement control and global cognition rather than an acute perinatal event. We consider that the underlying cerebellar involvement may be involved in his cognitive profile. In light of these results, it seems that sarcosinemia may lead to harmoniously small antenatal size and premature birth, followed by slow evolution of cognitive and neurological signs that affect daily living functions and social integration. Our case adds some additional evidence that sarcosinemia may be associated to neurodevelopmental disability.
Acknowledgements
We are grateful to Doctor Harvey Mudd for the invaluable help and discussion.
Appendix Relevant Conflicts of Interest/Financial Disclosures
Nothing to report. SB has been involved as main study coordinator in a clinical trial of memantine in Alzheimer’s disease.
Footnotes
Competing interests: None declared
References
- Bar-Joseph I, Pras E, Reznik-Wolf H, Marek-Yagel D, Abu-Horvitz A, Dushnitzky M, Goldstein N, Rienstein S, Dekel M, Pode-Shakked B, Zlotnik J, Benarrosh A, Gillery P, Hofliger N, Auray-Blais C, Garnotel R, Anikster Y (2012 Jul 24) Mutations in the sarcosine dehydrogenase gene in patients with sarcosinemia. Hum Genet (online first) [DOI] [PubMed]
- Botez-Marquard T, Léveillé J, Botez MI. Neuropsychological functioning in unilateral cerebellar damage. Can J Neurol Sci. 1994;21(4):353–357. doi: 10.1017/s0317167100040956. [DOI] [PubMed] [Google Scholar]
- Christensen E, Brandt NJ, Rosenberg T. Sarcosinaemia in a patient with Usher syndrome. J Inherit Metab Dis. 1989;12(4):487–488. doi: 10.1007/BF01802049. [DOI] [PubMed] [Google Scholar]
- Gerritsen T, Waisman HA. Hypersarcosinaemia: an inborn error of metabolism. N Eng J Med. 1966;275(2):66–69. doi: 10.1056/NEJM196607142750202. [DOI] [PubMed] [Google Scholar]
- Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46(2):73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees S, Inder T. Fetal and neonatal origins of altered brain development. Early Human Develop. 2005;81:753–761. doi: 10.1016/j.earlhumdev.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998;121(Pt 4):561–579. doi: 10.1093/brain/121.4.561. [DOI] [PubMed] [Google Scholar]
- Smith KE, Borden LA, Hartig PR, Branchek T, Weinshank RL. Cloning and expression of a glycine transporter reveal colocalization with NMDA receptors. Neuron. 1992;8(5):927–935. doi: 10.1016/0896-6273(92)90207-T. [DOI] [PubMed] [Google Scholar]
- van Sprang FJ, Duran M, Scholten HG, Wadman SK. A patient with sarcosinaemia. J Inherit Metab Dis. 1986;9(4):404–405. doi: 10.1007/BF01800497. [DOI] [PubMed] [Google Scholar]