

Abstract
Objective: Autosomal-recessive pyridox(am)ine phosphate oxidase (PNPO) deficiency causes pyridoxal-5-phosphate (PLP)-dependent epilepsy. We describe partial PNPO deficiency with a transient response to pyridoxine (B6).
Methods: CSF neurotransmitter metabolites, PLP, and amino acids were analyzed while the patient was receiving pyridoxine. PNPO gene sequencing was performed by standard techniques.
Results: A full-term 3,220 g male with refractory neonatal seizures became seizure free for 6 weeks on pyridoxine (B6). Breakthrough seizures followed. These stopped upon the first dose of PLP although episodes occurred as a dose became due. An unidentified peak was detected on the chromatographic system used to measure CSF PLP. PNPO gene sequencing identified a homozygous mutation in a highly conserved area in exon 3: c.352G>A p.G118R, predicting substitution of arginine for glycine. At age 28 months the child has hypotonia and developmental delay, both mild in severity.
Conclusions: Transient pyridoxine responsiveness may be seen in partial PNPO deficiency. A CSF metabolite peak, likely pyridoxine phosphate, is identifiable in patients with PNPO deficiency who are taking supplemental pyridoxine. Partial B6 responsiveness is an indication for possible PNPO deficiency and trial of PLP.
Introduction
Pyridoxine (B6)-dependent epilepsy was first described in 1954 (Hunt et al. 1954). The biological defect in this syndrome was recently identified as antiquitin mutations resulting in alpha aminoadipic semialdehyde (AASA) dehydrogenase deficiency (Mills et al. 2006). This enzyme is in the lysine degradation pathway and its deficiency leads to accumulation of intermediates that include pipecolic acid, AASA, and piperideine 6′-carboxylate. The latter sequesters pyridoxal 5′-phophate (PLP), leading to PLP deficiency within the central nervous system.
Pyridoxine (B6) is not itself an active cofactor. It is converted, together with the pyridoxal and pyridoxamine vitamers, to the active PLP via phosphorylation through the action of a kinase followed by oxidation by pyridox(am)ine 5′-phosphate oxidase (PNPO) (Surtees et al. 2006). Autosomal-recessive PNPO deficiency leads to a deficiency of PLP and subsequently to PLP-dependent epilepsy that, in general, is not thought to respond to pyridoxine (Mills et al. 2005). Without rapid detection and specific intervention, this causes catastrophic neonatal encephalopathy. In this report, we describe partial PNPO deficiency in an infant initially suspected to have pyridoxine responsiveness.
Methods
CSF neurotransmitter metabolites, pyridoxal-5-phosphate, and amino acid quantification. PNPO gene sequencing was performed by standard techniques on the proband followed by the parents.
Results
A full-term 3,220 g newborn male delivered following an uncomplicated gestational course and maternal labor presented with irritability, inconsolable crying, erratic eye movements with eyelid twitching, and grunting sounds 12 h following birth. EEG showed bilateral sharp discharges and episodic background suppression, although without a constant temporal correlation with the clinical symptoms. MRI was unremarkable. Following trials of phenobarbital and levetiracetam, the patient became seizure free for 6 weeks on pyridoxine. Breakthrough tonic seizures then occurred at 3.5 months of age, followed by myoclonic seizures, leading to hospitalization and trials of topiramate and prednisolone which were ineffective. EEG monitoring showed intermittent paroxysms of diffuse sharp electrographic activity associated variably with clinical facial grimacing, abnormal eye movements, and erratic multifocal myoclonias of the extremities which sometimes resembled thrashing type movements (Fig. 1). These were separated by periods of relative background suppression, accompanied by an initial tonic spasm and then quieting until the next paroxysm. This sequence persisted 25 min until intravenous lorazepam administration led to temporary resolution. The seizures stopped upon the first dose of PLP, but breakthrough events occurred as a dose became due. The patient achieved complete seizure control on PLP 10 mg/kg/dose given every 6 h.
Fig. 1.
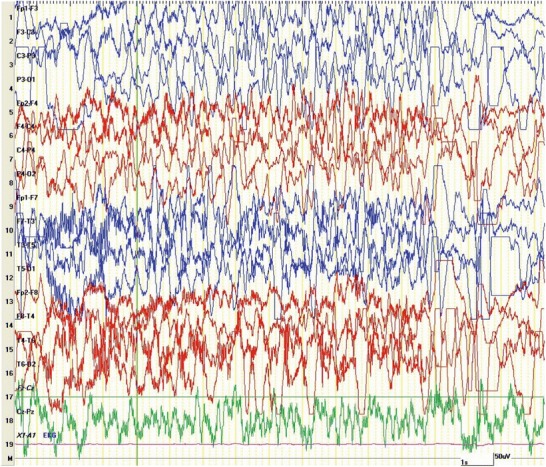
EEG of patient with PNPO deficiency, age 5 months, showing paroxysms of diffuse spike-wave discharges accompanied by multifocal myoclonic and erratic movements. Settings: 20 s epoch, sens 10 uV/mL, tc 0.1 s, HFF 70 Hz (Reproduced with permission from Alduligan and Pearl 2012)
CSF was sent for neurotransmitter metabolites, PLP, and amino acid analysis during the patient’s hospitalization with concomitant pyridoxine therapy. CSF PLP level was 23 nmol/L (normal range of 23–64 nmol/L). CSF amino acids showed a slight increase in threonine. Neurotransmitter metabolites were normal. An additional, previously unidentified, peak was detected on the chromatographic system used to measure PLP.
Subsequent PNPO gene sequencing identified a homozygous mutation in a highly conserved area in exon 3: c.352G>A p.G118R, predicting a substitution of arginine for glycine. Both parents were identified as heterozygous for this mutation. The patient has had sustained seizure control and developmental progress with ongoing dosing of PLP 10 mg/kg/dose administered four times daily with follow-up out to 2 years of age. Breakthrough clinical events tend to appear with intervals approaching 7–8 h after a prior dose, and examination shows mild symmetrical hypotonia and language delay. Developmental assessment at age 28 months indicated these levels: gross motor 21–24 months, fine motor 20–22 months, cognitive 20 months with scatter to 27 months, social-emotional 21–24 months, receptive language 24–28 months, expressive language 20–23 months, and self-help skills 21–24 months. His overall vocabulary is approximately 40 words, with the ability to place a few words together. However, much of his speech is repetition.
Discussion
The neonatal onset neurotransmitter disorders include both pyridoxine and PLP-responsive epilepsies (Pearl 2009). PLP, the biologically active form of B6, is a cofactor in approximately 120 enzymatic reactions, many of which involve amino acid and neurotransmitter metabolic pathways (Stockler et al. 2011). PNPO deficiency represents an epileptic encephalopathy resistant to pyridoxine but responsive to PLP.
The PNPO mutation identified, c.352G>A, has not been reported before. It is predicted by Polyphen to be probably damaging and by SIFT to be damaging. Our proband demonstrated an initial response in seizure control to oral pyridoxine dosing following refractoriness to standard antiepileptic drugs. When this control was lost after 6 weeks, CSF analysis was undertaken for neurotransmitter metabolite and PLP analysis. An unidentified compound was detected on the fluorescence chromatogram which has been noted in other patients with PNPO deficiency supplemented with pyridoxine. This peak is not detected in CSF samples derived from individuals with alpha-AASA dehydrogenase deficiency while on pyridoxine supplementation and is not the same peak seen in the chromatograms of folinic acid-responsive patients. We suspect this represents pyridoxine phosphate as this compound has been shown to accumulate in plasma in the absence of the PNPO (Footitt et al. 2012).
The seizure semiology in our patient is notably similar to the recently reported signs in four patients with pyridoxine-responsive epilepsy and one patient with PNPO deficiency (Schmitt et al. 2010). The features of neonatal and infantile irritability, inconsolable crying, facial grimacing, abnormal eye movements, eye twitching, multifocal myoclonias and erratic movements, and tonic seizures are characteristic of both diagnoses. The intermittent periods of EEG background suppression in our patient are remarkably similar to the PNPO-deficient patient in that series, as is the clinical observation of breakthrough events approximately 7 h after the last dose of PLP. The inconstant ictal EEG findings, absence of consistent clinical correlation with otherwise nonspecific paroxysmal EEG changes, and difficulty distinguishing interictal from ictal EEG background reported in both pyridoxine-responsive epilepsy and PNPO deficiency were also seen in our patient.
Patients without a defect in PNPO who receive pyridoxine generally have high-normal to above normal CSF PLP levels (KH – personal observation). The borderline low CSF PLP seen in our patient, while on pyridoxine, may indicate that the c.352G>A p.G118R mutation is “leaky” and allows for some oxidation of pyridoxine phosphate. This may explain the partial clinical response to pyridoxine. The pyridoxine dose was not increased, and it is plausible that a static pyridoxine dose was not sufficient to maintain efficacy as the child grew. Enzyme studies of the expressed mutant protein would be required to verify this hypothesis.
Our patient demonstrates that an initial response to pyridoxine that fails to materialize into permanent seizure control may be an indication of an underlying PNPO deficiency. In this situation, a trial with pyridoxal-5-phosphate is warranted.
Synopsis
We describe a novel case of partial pyridox(am)ine phosphate oxidase (PNPO) deficiency and pyridoxal-5-phosphate-dependent epilepsy with a transient response to pyridoxine (B6) treatment.
Footnotes
Competing interests: None declared
References
- Alduligan M, Pearl PL. Electroencephalography in metabolic epilepsies. In: Pearl PL, editor. Inherited metabolic epilepsies. New York: Demos Pubs; 2012. [Google Scholar]
- Footitt EJ, Clayton PT, Mills K et al (2012) Measurement of plasma B(6) vitamer profiles in children with inborn errors of vitamin B(6) metabolism using an LC-MS/MS method. J Inherit Metab Dis (2012) [Epub ahead of print] [DOI] [PubMed]
- Hunt AD, Jr, Stokes JJ, McCrory WW, Stroud HH. Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics. 1954;13:140–145. [PubMed] [Google Scholar]
- Mills PB, Surtees RA, Champion MP, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077–86. doi: 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307–309. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- Pearl PL. New treatment paradigms in neonatal seizures. J Inherit Metab Dis. 2009;32:204–213. doi: 10.1007/s10545-009-1045-8. [DOI] [PubMed] [Google Scholar]
- Schmitt B, Baumgartner M, Mills PB, et al. Seizures and paroxysmal events: symptoms pointing to the diagnosis of pyridoxine-dependent epilepsy and pyridoxine phosphate oxidase deficiency. Dev Med Child Neurol. 2010;52:e133–e142. doi: 10.1111/j.1469-8749.2010.03660.x. [DOI] [PubMed] [Google Scholar]
- Stockler S, Plecko B, Gospe SMJR, et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104:48–60. doi: 10.1016/j.ymgme.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Surtees R, Mills P, Clayton P. Inborn errors affecting vitamin B6 metabolism. Future Neurol. 2006;1(5):615–520. doi: 10.2217/14796708.1.5.615. [DOI] [Google Scholar]