

Abstract
Gaucher disease (GD) is an autosomal recessive disorder resulting from glucocerebrosidase (GC) deficiency due to mutations in the gene (GBA) coding for this enzyme. We have developed a strategy for analyzing the entire GBA coding region and applied this strategy to 48 unrelated Brazilian patients with GD. We used long-range PCR, genotyping based on the Taqman® assay, nested PCR, and direct DNA sequencing to define changes in the gene. We report here seven novel mutations that are likely to be harmful: S125N (c.491G>A), F213L (c.756T>G), P245T (c.850C>A), W378C (c.1251G>C), D399H (c.1312G>C), 982-983insTGC (c.980_982dupTGC), and IVS10+1G>T (c.1505+1G>T). The last alteration was found as a complex allele together with a L461P mutation. We also identified 24 different mutations previously reported by others. G377S was the third most frequent mutation among the patients included in this study, after N370S and L444P. Therefore, this mutation needs be included in preliminary screens of Brazilian GD patients. The identification of mutant GBA alleles is crucial for increasing knowledge of the GBA mutation spectrum and for better understanding of the molecular basis of GD.
Introduction
Gaucher disease (GD) is the most common lysosomal storage disorder and results from an inborn deficiency of the enzyme glucocerebrosidase (GC; EC 3.2.1.45; also known as acid β-glucosidase) (Beutler and Grabowski 2001). This enzyme is responsible for glycosphingolipid glucocerebroside (glucosylceramide) degradation. Enzyme deficiency leads to the accumulation of undegraded substrate, mainly within cells of the monocyte/macrophage lineage, and this is responsible for the clinical manifestations of the disease. Three types of GD are distinguished based on neurological involvement. GD is an autosomal recessive disorder caused by mutations in the glucocerebrosidase gene or, rarely, by mutations in the GC activator protein, saposin C (Beutler and Grabowski 2001).
The gene encoding GC (GBA; GenBank accession # J03059) is located on chromosome 1q21 and spans 7.6 kb of genomic DNA divided into 11 exons. In addition to the functional gene, a highly homologous pseudogene sequence (GBAP; GenBank accession # J03060) is located 16 kb downstream (Horowitz et al. 1989; Winfield et al. 1997). The gene and pseudogene are in the same orientation and have 96 % exonic sequence homology (Horowitz et al. 1989; Sidransky 2004). To date, more than 350 mutations have been reported in GBA. These include missense and nonsense mutations, small insertions or deletions that lead to frameshifts or in-frame alterations, splice junction mutations, and complex alleles carrying two or more mutations (Hruska et al. 2008). The frequencies of specific mutant alleles vary in different populations. In patients of Ashkenazi Jewish ancestry, four mutations account for nearly 90 % of the disease alleles. These mutations are N370S (c.1226A>G), L444P (c.1448T>C), 84insG (c.84dupG), and IVS2+1G>A (c.115+1G>A). Among non-Jewish patients, these mutations account for about 50–60 % of GD-associated mutations and there is a broad spectrum of other mutations (Grabowski and Horowitz 1997; Alfonso et al. 2007).
The diagnosis of GD is established by measuring GC activity in leukocytes from peripheral blood and/or fibroblasts from skin biopsies (Beutler and Grabowski 2001). Molecular analysis complements this biochemical assay. Our group has been involved in the biochemical diagnosis of Brazilian patients with GD for the last 20 years (Michelin et al. 2005). We have also introduced screening for the common mutations (N370S, L444P, 84insG, and IVS2+1G>A). As expected, this approach is able to detect roughly half the mutant alleles in our sample population, and just over 60 % of patient genotypes. Hence, a more comprehensive scheme is desirable in order to be able to identify the remaining mutant alleles.
We have therefore designed a strategy for analyzing the entire GBA coding region and applied this approach to 48 unrelated Brazilian patients with GD among 128 patients referred to our laboratory and previously confirmed by biochemical analysis. We describe here seven novel mutations associated with GD, as well as other rare GBA mutations.
Materials and Methods
Patients
In this study, we examined 48 unrelated non-Jewish GD patients from different regions of Brazil. There were 24 males and 24 females. Ages at diagnosis, when available, ranged from 2 months to 57 years. The inclusion criteria were (1) low GC activity in leukocytes and/or fibroblasts (Michelin et al. 2005) and (2) at least one unidentified disease-causing allele following screening for the common mutations (N370S, L444P, 84insG, and IVS2+1G>A). The study was approved by our hospital ethics committee.
In order to confirm that we were dealing with meaningful sequence alterations, we also sequenced GBA in DNA from 104 (208 alleles) healthy Brazilian subjects (52 males and 52 females) to rule out the possibility that some of the novel variants identified were simply polymorphisms.
Sample Collection and DNA Isolation
Blood samples (5 mL) were collected in EDTA, and genomic DNA was isolated from peripheral blood leukocytes as described (Miller et al. 1988) and kept at –20 °C. DNA was quantified with a fluorescence-based kit (Quant-ItTM dsDNA BR Assay kit; Invitrogen, Carlsbad, CA, USA) in a QubitTM fluorometer (Invitrogen).
Amplification of the Entire GBA Gene
Long-range polymerase chain reaction (PCR) was used to selectively amplify the functional GBA gene using primers GBALF (5′ CGACTTTACAAACCTCCCTG 3′) and GBALR (5′ CCAGATCCTATCTGTGCTGG 3′); this generated a fragment of 7765 bp. The long-range PCR reaction was performed in final volumes of 25 μL containing 12.5 ng genomic DNA, 200 μM of each dNTP, 0.2 μM of each primer (forward and reverse), 60 mM of Tris-SO4 (pH 9.1), 18 mM of (NH4)2SO4, 1.7 mM of MgSO4, and 1 μL of Elongase® Enzyme Mix (Invitrogen, Carlsbad, CA, USA). Cycling conditions were initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s and elongation at 68 °C for 8 min, with final extension at 68 °C for 10 min.
Screening of Common Mutations
Mutations N370S and L444P were screened by genotyping based on TaqMan® PCR (Applied Biosystems, Foster City, CA, USA). Primers and probes were designed with Primer Express® software version 3.0 (Applied Biosystems), and the primer sequences are shown in Table 1. PCR reactions were performed in final volume of 12 μL containing 0.5 μL of the long-range PCR product, 0.3 μL of specific TaqMan assay medium and 6 μL of 2x PCR Genotyping Master Mix (Applied Biosystems). Amplification included an initial step at 50 °C for 2 min (activation of the AmpErase UNG function), AmpliTaq® Gold activation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing extension at 60°C for 1 min. The allelic discrimination step was performed at 60°C for 1 min. PCR products were analyzed by allelic discrimination plot with Sequence Detection System software version 1.2.1 in an ABI PRISM® 7500 Sequence Detector System (Applied Biosystems).
Table 1.
Sequences of the primers and probes used to identify frequent mutations
| Mutation | Primer sequence (5´ > 3´) | Probe sequence (5´ > 3´) |
|---|---|---|
| N370S | Forward | Normal |
| GCCTTTGTCTCTTTGCCTTTGTC | TTACCCTAGAACCTCCTG – VIC | |
| Reverse | Mutant | |
| CCAGCCGACCACATGGTA | ACCCTAGAGCCTCCTG – FAM | |
| L444P | Forward | Normal |
| CTGAGGGCTCCCAGAGAGT | CTGCGTCCAGGTCGT – VIC | |
| Reverse | Mutant | |
| GCCATCGGGATGCATCAGT | TGCGTCCGGGTCGT – FAM |
Mutation 84insG was screened by Amplification Refractory Mutation System-PCR (ARMS-PCR). Two PCR reactions were performed for each sample using different specific primers to discriminate between wild-type and mutant alleles. Primers GAU-84GGWRT (5′ GCATCATGGCTGGCAGCCTCACAGGACTGC 3′) and GAU-2R (5′ GCCCAGGCAACAGAGTAAGACTCTGTTTCA 3′) were used to amplify the wild-type allele, and GAU-84GGMTF (5′ GCATCATGGCTGGCAGCCTCACAGGACTGG 3′) and GAU-2R for the mutant allele. These reactions generated a fragment of 255 bp when the allele sequence was complementary to the primer sequence. Each PCR reaction was performed in a total volume of 25 μL, containing 200 ng of genomic DNA, 200 μM of each dNTP, 1 μM of each primer (forward and reverse), 20 mM of Tris (pH 8.4), 50 mM of KCl, 2.5 mM of MgCl2, and 1.25 U of Taq DNA polymerase. Amplification conditions were 96 °C for 5 min, followed by 13 cycles of 96 °C for 45 s, then 45 s at 70 °C −0.5 °C/cycle, and 72 °C for 45 s, then 22 cycles of 96 °C for 45 s, 64 °C for 45 s, and 72 °C for 45 s, with final extension at 72 °C for 5 min. Amplified products were resolved by electrophoresis on 2 % (w/v) agarose gels and visualized under UV light.
Mutation IVS2+1G>A was screened by a PCR-Restriction Fragment Length Polymorphism (PCR-RFLP) strategy using primers GAU-84GGWRT and GAU-2R (sequences shown above) that generated a fragment of 255 bp. The PCR reaction was performed in a total volume of 25 μL, containing 200 ng of genomic DNA, 200 μM of each dNTP, 1 μM of each primer (forward and reverse), 20 mM of Tris (pH 8.4), 50 mM of KCl, 2.5 mM of MgCl2, and 1.25 U of Taq DNA polymerase. The amplification protocol was initial denaturation at 96 °C for 5 min, 35 cycles of denaturation at 96 °C for 45 s, annealing at 68 °C for 45 s, and extension at 72 °C for 45 s, followed by final extension at 72 °C for 10 min. Ten μL of PCR product was digested in a total volume of 15 μL containing 20 mM of Tris-acetate, 50 mM of potassium acetate, 10 mM of magnesium acetate, 1 mM of dithiothreitol (pH 7.9), and 1 U of HphI (New England Biolabs, Ipswich, MA, USA). The reaction was then placed at 37 °C and left overnight, after which digestion products were resolved by electrophoresis on 3 % (w/v) agarose gels and visualized under UV light. Digestion of the PCR product of the normal allele generates fragments of 141, 72, and 42 bp, while digestion of the PCR product of the mutant allele produces fragments of 213 and 42 bp, because the IVS2+1G>A mutation removes an HphI site.
PCR Amplification and Direct DNA Sequencing
Coding sequences and flanking regions (exons 1 to 11) were amplified by PCR using long-range PCR products as templates. The GBA coding region was divided into 10 different amplicons, with exons 10 and 11 analyzed together. PCR reactions were performed in total volumes of 25 μL, containing 0.5 μL of long-range PCR product, 200 μM of each dNTP, 0.2 μM of each primer (forward and reverse), 20 mM of Tris (pH 8.4), 50 mM of KCl, 2.5 mM of MgCl2, and 1.25 U of Taq DNA polymerase. The amplification protocol was initial denaturation at 94 °C for 10 min, 30 cycles of denaturation at 94 °C for 30 s, annealing at 57–62 °C for 30 s, and extension at 72 °C for 30 s, followed by final extension at 72 °C for 10 min. Each PCR product was verified by electrophoresis on a 1.5 % (w/v) agarose gel and visualization under UV light. Specific annealing temperatures and primer sequences are given in Table 2.
Table 2.
Primers used to amplify and sequence the GBA gene
| Exons and flanking regions | Primer sequence (5´ > 3´) | Tannealing
(0C) |
Length (bp) |
|
|---|---|---|---|---|
| Forward | Reverse | |||
| 1 | CCTAGTGCCTATAGCTAAGG | CTGGATTCAAAGAGAGTCTG | 57 | 236 |
| 2 | GTCCTAATGAATGTGGGAGACC | CTTACTGGAAGGCTACCAAAGG | 61 | 286 |
| 3 | GTTCAGTCTCTCCTAGCAGATG | GGAAACTCCATGGTGATCAC | 61 | 353 |
| 4 | GTCCTCCTAGAGGTAAATGGTG | GCAGAGTGAGATTCTGCCTC | 61 | 316 |
| 5 | GATAAGCAGAGTCCCATACTCTC | CTGTACAAGCAGACCTACCCTAC | 62 | 281 |
| 6 | CTAATGGCTGAACCGGATG | GGAAGTGGAACTAGGTTGAGG | 62 | 346 |
| 7 | CAAAGTGCTGGGATTACAGG | CTCTAAGTTTGGGAGCCAGTC | 62 | 404 |
| 8 | CTAGTTGCATTCTTCCCGTC | GCTTCTGTCAGTCTTTGGTG | 63 | 407 |
| 9 | CTCCCACATGTGACCCTTAC | CTCGTGGTGTAGAGTGATGTAAG | 61 | 329 |
| 10 and 11 | GTGGGTGACTTCTTAGATGAGG | CTTTAGTCACAGACAGCGTGTG | 62 | 473 |
Amplicons were purified using 2.5 U of Exonuclease I (USB, Cleveland, OH, USA) and 0.25 U of Shrimp Alkaline Phosphatase (USB, Cleveland, OH, USA). Direct DNA sequencing was performed with a BigDye® Terminator Cycle Sequencing kit v. 3.1 (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions, and sequences were analyzed with DNA Sequencing Analysis software v. 5.2 (Applied Biosystems) in an ABI PRISM® 3130xl Genetic Analyzer. All identified mutations were confirmed by sequencing an independent DNA sample with forward and reverse primers.
Isolation of Total RNA, cDNA Synthesis, and PCR Amplification
Total RNA was isolated from peripheral blood leukocytes using a LeukoLOCKTM Total RNA Isolation kit (Applied Biosystems). RNA was quantified using a fluorescence-based assay (Quant-ItTM RNA Assay kit) in a QubitTM fluorometer.
RNA samples of 1 μg were used as templates for complementary DNA (cDNA) synthesis. Reverse transcription (RT) was performed with a High Capacity cDNA Reverse Transcription kit (Applied Biosystems) in a total volume of 15 μL. Following the RT reaction, PCR was performed using primers GAU-G10S (5′ CTGAACCCCGAAGGAGGACC 3′) and GAU-9/11R (5′ GGTTTTTCTACTCTCATGCA 3′), which generated a product of 988 bp. Five microliters of each RT reaction was used for the PCR in a total volume of 25 μL, containing 200 μM of each dNTP, 0.8 μM of each primer (forward and reverse), 20 mM of Tris (pH 8.4), 50 mM of KCl, 1.5 mM of MgCl2, and 1.25 U of Taq DNA polymerase. The amplification protocol was initial denaturation at 94 °C for 5 min, 30 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min, followed by final extension at 72 °C for 10 min. An aliquot of each PCR product was verified by electrophoresis on a 1.0 % (w/v) agarose gel and visualization under UV light. PCR products were purified and sequenced as described above.
Evaluation of Novel Mutations
Amino acid sequences of GC from 12 different species were compared by multiple alignment program in order to determine whether changes identified in their amino acid sequences alter conserved residues of GC. The GC sequences were searched using the protein database from the National Center for Biotechnology Information (NCBI). The amino acid sequences were aligned with ClustalW v. 2.0 using the FASTA format.
The novel mutations in the GBA coding region were analyzed using three web-based tools, PolyPhen (http://genetics.bwh.harvard.edu/pph) (Ramensky et al. 2002), SNPs3D (http://www.snps3d.org) (Yue and Moult 2006), and SIFT (http://sift.bii.a-star.edu.sg) (Kumar et al. 2009) in order to assess their potential pathogenicity.
Nomenclature of Mutations
The reference sequences used in our study were NM_000157 and NP_000148. Nucleotide sequences were numbered from the upstream initiator codon ATG. Amino acid numbers are those of the mature proteins after cleavage of the leader sequence. Following the current recommendation that all variants be described at the most basic level, the mutations discussed in the text are referred to by the traditional name of the corresponding allele to facilitate recognition, followed by the cDNA nomenclature in brackets (den Dunnen and Antonarakis 2000).
Results
Screening the whole GBA coding sequence in each of the 48 GD patients allowed us to identify 95.8 % of the mutant alleles, including seven novel sequence variants. These novel changes were not found among 208 alleles from normal individuals. Twenty-four rare mutations were also found among the mutant alleles studied. Data for these 31 variants are summarized in Table 3.
Table 3.
Alleles defined by this study. The novel sequence variants are shown in bold
| GBA mutationsa | cDNA nucleotide substitutionb | Exon | Proteinc | # of alleles |
|---|---|---|---|---|
| Substitutions | ||||
| R48Q | c.260G>A | 3 | p.Arg87Gln | 1 |
| R120Wd | c.475C>T | 5 | p.Arg159Trp | 3 |
| M123T | c.485T>C | 5 | p.Met162Thr | 1 |
| S125N | c.491G>A | 5 | p.Ser164Asn | 1 |
| R131C | c.508C>T | 5 | p.Arg170Cys | 1 |
| W179X | c.653G>A | 6 | p.Trp218X | 1 |
| N188Sd | c.680A>G | 6 | p.Asn227Ser | 1 |
| G202Rd | c.721G>A | 6 | p.Gly241Arg | 1 |
| F213L | c.756T>G | 6 | p.Phe252Leu | 1 |
| P245T | c.850C>A | 7 | p.Pro284Thr | 1 |
| H311R | c.1049A>G | 8 | p.His350Arg | 1 |
| Y313H | c.1054T>C | 8 | p.Tyr352His | 1 |
| E349K | c.1162G>A | 8 | p.Glu388Lys | 1 |
| R353W | c.1174C>T | 8 | p.Arg392Trp | 1 |
| N370S | c.1226A>G | 9 | p.Asn409Ser | 26 |
| G377S | c.1246G>A | 9 | p.Gly416Ser | 12 |
| W378C | c.1251G>C | 9 | p.Trp417Cys | 6 |
| N396T | c.1304A>T | 9 | p.Asn435Thr | 4 |
| V398I | c.1309G>A | 9 | p.Val437Ile | 6 |
| D399H | c.1312G>C | 9 | p.Asp438His | 1 |
| L444Pd | c.1448T>C | 10 | p.Leu483Pro | 2 |
| I489T | c.1583T>C | 11 | p.Ile528Thr | 7 |
| R496H | c.1604G>A | 11 | p.Arg535His | 1 |
| Insertions | ||||
| 84insG | c.84dupG | 2 | p.Leu29AlafsX18 | 1 |
| 982-983insTGC | c.980_982dupTGC | 7 | p.Leu327_Pro328insLeu | 1 |
| Deletions | ||||
| 413delC | c.413delC | 4 | p.Pro138LeufsX62 | 1 |
| 793delC | c.793delC | 7 | p.Gln265SerfsX5 | 2 |
| Complex alleles | ||||
| E326K + L444Pd | c.1093G>A | 8 | p.Glu365Lys | 1 |
| c.1448T>C | 10 | p.Leu483Pro | ||
| M361I + N370S | c.1200G>A | 8 | p.Met400Ile | 1 |
| c.1226A>G | 9 | p.Asn409Ser | ||
| c.1448T>C | 10 | p.Leu483Pro | ||
| RecNciI | c.1483G>C | 10 | p.Ala495Pro | 3 |
| c.1497G>C | 10 | p.Val499Val | ||
| L461Pd + IVS10+1G>T | c.1499T>C | 10 | p.Leu500Pro | 1 |
| c.1505+1G>T | p.Lys464_Arg502del |
a GBA mutations are named according to www.hgvs.org/mutnomen
bNucleotides are numbered from the A of the first ATG
cAmino acid designations (“p.”) are based on the primary GBA translation product, including the 39-residue signal peptide
dThese alterations correspond to the normal pseudogene sequence
The novel alterations consist of five missense changes [S125N (c.491G>A), F213L (c.756T>G), P245T (c.850C>A), W378C (c.1251G>C), and D399H (c.1312G>C)], one in-frame insertion [982-983insTGC (c.980_982dupTGC)], and a splicing mutation found in a complex allele [L461P + IVS10+1G>T]. These findings are described in more detail below.
The first novel sequence alteration is caused by a G to A change at position 491 of the cDNA (exon 5 of GBA) and leads to a serine to asparagine substitution at residue 125 of the protein (S125N). This mutation was found in a male type 1 GD patient from the south of Brazil who was diagnosed at 56 years of age and carries the N370S mutation on the other chromosome. Abdominal pain, splenomegaly, and chronic bone pain are among the clinical features of this patient.
The GBA of another type 1 GD patient has a T to G transversion at base 756 of the cDNA (exon 6 of the gene) predicting a phenylalanine to leucine substitution at residue 213 of the protein (F213L). The N370S mutation was also present in this compound heterozygous female type 1 GD patient. She is from the Southeast of Brazil and was diagnosed when she was 26 years old. Clinical features include hepatosplenomegaly and bone pain.
Two novel sequence variants are in exon 7. The first is a C to A change at position 850 of the cDNA predicting a proline to threonine substitution at amino acid 245 of protein (P245T). This was identified in a male type 3 GD patient from the southeast region of Brazil who is also heterozygous for RecNciI (c.1448T>C + c.1483G>C + c.1497G>C). This patient is more severely affected, with neurological involvement. The other alteration in exon 7 is an in-frame insertion of three nucleotides (TGC) that should insert an additional leucine residue between positions 327 and 328 of the mature protein (Fig. 1a). This mutation is referred to as 982-983insTGC and was found in a male type 1 patient from the southeast of Brazil diagnosed at 31 years of age. The other mutant allele in this patient is N370S, and his clinical features include hepatosplenomegaly.
Fig. 1.
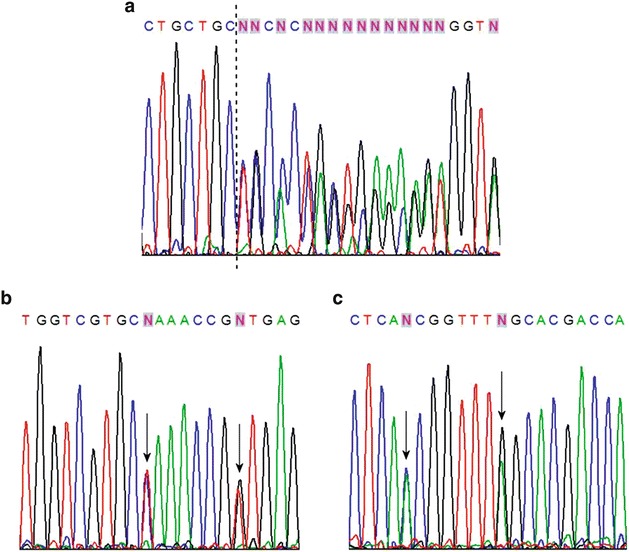
Direct DNA sequencing of the GBA gene. (a) Direct sequencing of part of exon 7 from the forward primer. The dashed line shows where three nucleotides (TGC) have been inserted, creating the 982-983insTGC mutation. (b) Direct sequencing of part of exon 10 from the forward primer. The first arrow indicates the T to C substitution (c.1499T>C) in the L461P mutation, while the second arrow points to the G to T substitution in the IVS10+1G>T variant. (c) Direct sequencing of part of exon 10 from the reverse primer. The first arrow indicates the C to A substitution in IVS10+1G>T, while the second arrow points to the A to G substitution in L461P
Two novel G to C transversions are in exon 9. The first is at base 1251 of the GBA cDNA and causes a tryptophan to cysteine substitution at residue 378 of the mature protein (W378C). This alteration was found in unrelated alleles in six type 1 GD patients from the north and northeast regions of Brazil. One of these patients carries the G377S (c.1246G>A) mutation on the other chromosome, while the remaining five patients are compound heterozygotes [N370S] + [W378C]. All six patients have typical mild forms of the disease consistent with type 1 disease. The other G to C transversion is located at position 1312 and is responsible for an aspartate to histidine substitution at residue 399 of the protein (D399H). This mutation was found in a type 1 GD patient diagnosed at 45 years of age who is also heterozygous for the N370S mutation. The most characteristic finding in this patient was extreme hepatosplenomegaly.
The remaining novel mutation involves a G to T transversion in the splice donor site at the first nucleotide of intron 10 (Fig. 1b and c). This mutation, named IVS10+1G>T, removes the conserved splice donor sequence and results in skipping of exon 10. RT-PCR amplification of the GBA cDNA from this patient revealed an 871 bp fragment in addition to the expected 988 bp fragment (from the other allele) (data not shown). This smaller product was sequenced and shown to lack exon 10, presumably due to loss of the splice donor site at the end of exon 10 (Fig. 2b). A control cDNA used at the same time yielded a normal splicing pattern and a fragment of the expected length (Fig. 2a). Schematic representations of the normal and aberrant splicing patterns are shown in Fig. 2c and d, respectively. This variant was found in a female type 1 patient from the south of Brazil diagnosed at 3 years of age. N370S and L461P (c.1499T>C) were also present in this patient. As DNA from her parents was also available, we were able to show that the N370S mutation was of paternal origin, while L461P and IVS10+1G>T were of maternal origin.
Fig. 2.
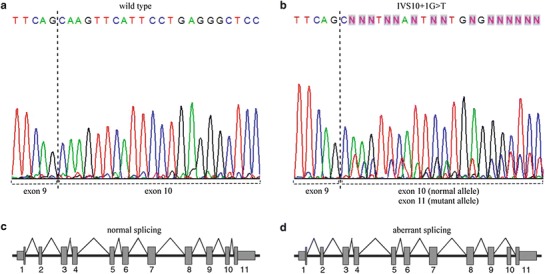
Direct sequencing of GBA cDNA. (a) Wild-type alleles sequenced from the forward primer. The dashed line indicates the boundary between exons 9 and 10. (b) Normal and mutant alleles from a GD patient carrying IVS10+1G>T, sequenced from the forward primer. The dashed lines indicate the boundary between exons 9 and 10 in the normal allele and between exons 9 and 11 in the mutant allele, respectively. Schematic diagrams of (c) the normal splicing pattern and (d) the aberrant splicing pattern due to the IVS10+1G>T mutation
In silico evaluation of the five novel sequence variants using PolyPhen, SNPs3D, and SIFT predicted defective proteins for S125N, W378C, and D399H. However, the three bioinformatics tools differed in their predictions for the F213L and P245T variants. PolyPhen predicted that they would be deleterious whereas according to SNPs3D and SIFT, they were non-deleterious.
We also estimated the frequencies of the common previously reported mutations taking into account all 128 patients referred to our laboratory. As expected, the most frequent mutation was N370S, found in 113 alleles (44.1 %); the second most frequent mutation in our cohort, L444P, was found in 73 alleles (28.5 %); and G377S, the third most frequent mutation, accounted for 12 mutant alleles (11.1 %). This was followed by I489T (c.1583T>C) seven alleles (6.5 %), W378C and V398I (c.1309G>A) six alleles each (5.6 %), RecNciI three alleles (2.8 %), IVS2+1G>A two alleles (1.9 %), and [E326K + L444P] one allele (0.9 %).
The genotypes of the 48 GD patients examined in this study are shown in Table 4. Genotypes involving only combinations of the common mutations were excluded.
Table 4.
Frequencies of the identified genotypes
| Genotype | # of patients |
|---|---|
| [N370S] + [W378C] | 5 |
| [G377S] + [G377S] | 4 |
| [V398I] + [V398I] | 3 |
| [N370S] + [I489T] | 3 |
| [R120W] + [N370S] | 3 |
| [793delC] + [N370S] | 2 |
| [N370S] + [L461P + IVS10+1G>T] | 1 |
| [Y313H] + [L444P] | 1 |
| [G377S] + [I489T] | 1 |
| [R353W] + [RecNciI] | 1 |
| [F213L] + [N370S] | 1 |
| [W179X] + [M361I + N370S] | 1 |
| [N188S] + [RecNciI] | 1 |
| [M123T] + [E349K] | 1 |
| [G377S] + [W378C] | 1 |
| [S125N] + [N370S] | 1 |
| [P245T] + [RecNciI] | 1 |
| [N396T] + [N396T] | 1 |
| [N370S] + [D399H] | 1 |
| [413delC] + [N370S] | 1 |
| [I489T] + [I489T] | 1 |
| [N370S] + [G377S] | 1 |
| [N396T] + [I489T] | 1 |
| [N396T] + [L444P] | 1 |
| [G202R] + [N370S] | 1 |
| [R131C] + [N370S] | 1 |
| [84insG] + [R496H] | 1 |
| [H311R] + [N370S] | 1 |
| [982-983insTGC] + [N370S] | 1 |
| [G377S] + [E326K + L444P] | 1 |
| [N370S] + [?] | 3 |
| [R48Q] + [?] | 1 |
| Total | 48 |
? denotes unidentified mutation
Discussion
The protocol described here allowed us to identify seven novel sequence variants of the GBA gene in patients with GD. These include five missense mutations (S125N, F213L, P245T, W378C, and D399H). We also found that the G377S mutation was common among the patients in this study, as previously reported by other Brazilian groups (Rozenberg et al. 2006; Sobreira et al. 2007). Therefore, G377S should be included in preliminary screening of Brazilian GD patients.
The S125N mutation involves the replacement of a polar uncharged amino acid with a hydroxyl group by an uncharged residue with an amide group containing a long side chain. Ser125 is conserved in several species from human to worms, arguing in favor of a role in enzyme function (Fig. 3). This mutation destroys a PvuII restriction site. The S125N mutation was previously reported in patients with Parkinson disease (Lesage et al. 2011); however, this sequence alteration has not been identified in GD patients to date.
Fig. 3.
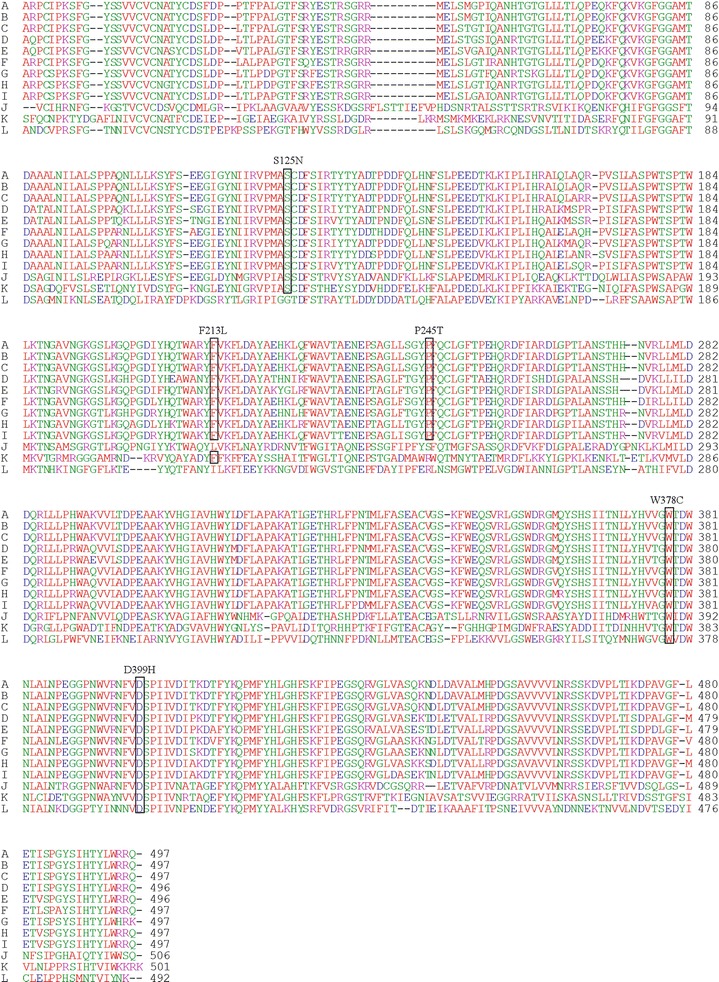
Alignment of the GC amino acid sequences of several organisms. (a) Human (Gene ID 2629); (b) chimpanzee (Gene ID 449571); (c) orangutan (Gene ID 100174563); (d) rat (Gene ID 684536); (e) mouse (Gene ID 14466); (f) dog (Gene ID 612206); (g) pig (Gene ID 449572); (h) ox (Gene ID 537087); (i) horse (Gene ID 100063514); (j) tick (Gene ID 8051161); (k) C. elegans (Gene ID 178535); (l) honeybee (Gene ID 409708). The boxes indicate conserved residues altered in novel missense mutations. The designation of each mutation is given above the appropriate box
The F213L mutation replaces a bulky aromatic amino acid by a residue containing an open aliphatic chain, which possibly changes the protein conformation. Although in silico evaluation predicts a non-deleterious effect on GC, this phenylalanine residue is conserved in several species, such as chimpanzee, orangutan, rat, mouse, dog, pig, ox, and horse (Fig. 3).
The other mutation, P245T, involves the replacement of a cyclic aliphatic structure by an open polar uncharged chain and would be expected to affect protein structure and/or activity. In silico evaluation of P245T predicted a non-deleterious effect on the mature protein. However, the proline residue is conserved in mammals such as chimpanzee, orangutan, rat, mouse, dog, pig, ox, and horse (Fig. 3).
The mutation in exon 9, W378C, involves the replacement of an amino acid with an indole side chain by an amino acid with a sulfhydryl group, which may affect protein conformation. In silico analysis predicted a deleterious effect. The tryptophan residue is highly conserved, indicating a considerable degree of structural and functional relevance (Fig. 3). This mutation creates an HpyCH4V restriction site within GBA.
D399H involves the replacement of an acidic amino acid by a basic one, which is again likely to affect protein structure and/or function. Asp399 is highly conserved, and in silico analysis predicts a deleterious effect of the mutation (Fig. 3). This mutation destroys a HindII restriction site.
Two other variants likely to have harmful effects are an insertion and a single base change at the first position of a splice donor site. Despite being an in-frame insertion, the insertion of an additional residue in the mature protein is likely to disturb normal folding and function. The point mutation in the first base of the splice donor site disrupts normal splicing, giving rise to exon 10 skipping, which yields a truncated protein of 458 amino acids (p.Lys464_Arg502del).
Most of the novel sequence alterations detected in this study were found in patients with the N370S mutation at the other locus. It appears that this allele when part of any genotype is able to protect patients from neurological involvement (Alfonso et al. 2001). Indeed, no neurological involvement was reported in any of our patients of this type. Moreover, neurological impairment is not seen in GD patients who carry N370S or 982-983insTGC, which further support a protective effect of the N370S mutation.
Interestingly, the G377S mutation was also found to be the third most frequent among GD patients in Portugal and Spain (Amaral et al. 1996). This alteration appears to be neuroprotective since homoallelic patients are essentially asymptomatic or have mild disease (type 1) (Amaral et al. 2000). However, other workers have detected type 3 patients with one copy of G377S (Beutler et al. 2005; Rozenberg et al. 2006). Thus we cannot rule out an allele-dose effect in which two copies of G377S generate sufficient residual enzymatic activity to prevent neurological involvement, whereas one copy combined with a severe second allele leads to type 3 GD (Rozenberg et al. 2006). Such a model predicts that our patient with [G377S] + [E326K + L444P] is likely to develop neurological symptoms, although no such symptoms have been described to date.
There remain undefined mutant alleles in our collection that can be explained by alterations outside the GBA coding region. Such changes would not be detected by our approach, nor would sequence alterations in the promoter region of the gene, or in untranslated or in noncoding regions. We are currently testing further approaches to overcome these limitations and unmask the remaining mutant alleles.
The vast majority of mutations identified have not been found in homoallelic form, especially the new variants. Ultimately, it may be more accurate to envision the associated phenotypes as forming a continuum, with the major distinction being the presence and degree of neurological involvement (Sidransky 2004; Hruska et al. 2008).
The identification of mutant alleles is crucial for advancing knowledge of the worldwide GBA mutation spectrum, and should contribute to a better understanding of the molecular basis of the disease. Such information should also help in establishing genotype-phenotype correlations as well as in genetic counseling and/or in customized molecular analyses for families at risk.
Acknowledgments
The authors are grateful to the patients and their families who kindly agreed to participate in this study. We also thank the physicians who referred patients to our laboratory and provided clinical information. The authors would also like to thank Thais Santa Rita for her laboratory technical assistance in this work. MS, JCC, RG, and MLSP were supported by CNPq, Brazil. HB was supported by CAPES, Brazil. The work was also supported by Brazilian Funding Agencies (CNPq, FAPERGS, and FIPE-HCPA) and by Genzyme do Brazil.
Synopsis
Novel and rare mutations in the GBA gene of patients with Gaucher disease.
Footnotes
Competing interests: None declared
References
- Alfonso P, Cenarro A, Perez-Calvo JI, et al. Mutation prevalence among 51 unrelated Spanish patients with Gaucher disease: identification of 11 novel mutations. Blood Cells Mol Dis. 2001;27:882–891. doi: 10.1006/bcmd.2001.0461. [DOI] [PubMed] [Google Scholar]
- Alfonso P, Aznarez S, Giralt M, et al. Mutation analysis and genotype/phenotype relationships of Gaucher disease patients in Spain. J Hum Genet. 2007;52:391–396. doi: 10.1007/s10038-007-0135-4. [DOI] [PubMed] [Google Scholar]
- Amaral O, Pinto E, Fortuna M, et al. Type 1 Gaucher disease: identification of N396T and prevalence of glucocerebrosidase mutations in the Portuguese. Hum Mutat. 1996;8:280–281. doi: 10.1002/(SICI)1098-1004(1996)8:3<280::AID-HUMU15>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Amaral O, Marcao A, Sa Miranda M, et al. Gaucher disease: expression and characterization of mild and severe acid beta-glucosidase mutations in Portuguese type 1 patients. Eur J Hum Genet. 2000;8:95–102. doi: 10.1038/sj.ejhg.5200422. [DOI] [PubMed] [Google Scholar]
- Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3635–3668. [Google Scholar]
- Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis. 2005;35:355–364. doi: 10.1016/j.bcmd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Grabowski GA, Horowitz M. Gaucher's disease: molecular, genetic and enzymological aspects. Baillieres Clin Haematol. 1997;10:635–656. doi: 10.1016/S0950-3536(97)80032-7. [DOI] [PubMed] [Google Scholar]
- Horowitz M, Wilder S, Horowitz Z, et al. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 1989;4:87–96. doi: 10.1016/0888-7543(89)90319-4. [DOI] [PubMed] [Google Scholar]
- Hruska KS, LaMarca ME, Scott CR, et al. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA) Hum Mutat. 2008;29:567–583. doi: 10.1002/humu.20676. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Condroyer C, et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson´s disease. Hum Mol Genet. 2011;20:202–210. doi: 10.1093/hmg/ddq454. [DOI] [PubMed] [Google Scholar]
- Michelin K, Wajner A, de Souza FT, et al. Application of a comprehensive protocol for the identification of Gaucher disease in Brazil. Am J Med Genet. 2005;136:58–62. doi: 10.1002/ajmg.a.30787. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenberg R, Araujo FT, Fox DC, et al. High frequency of mutation G377S in Brazilian type 3 Gaucher disease patients. Braz J Med Biol Res. 2006;39:1171–1179. doi: 10.1590/S0100-879X2006000900004. [DOI] [PubMed] [Google Scholar]
- Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83:6–15. doi: 10.1016/j.ymgme.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Sobreira E, Pires RF, Cizmarik M, Grabowski GA. Phenotypic and genotypic heterogeneity in Gaucher disease type 1: a comparison between Brazil and the rest of the world. Mol Genet Metab. 2007;90:81–86. doi: 10.1016/j.ymgme.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Winfield SL, Tayebi N, Martin BM, et al. Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: implications for Gaucher disease. Genome Res. 1997;7:1020–1026. doi: 10.1101/gr.7.10.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Moult J. Identification and analysis of deleterious human SNPs. J Mol Biol. 2006;356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]