

Abstract
Deficiency of ornithine-δ-aminotransferase (OAT) in humans results in gyrate atrophy. Early diagnosis may allow initiation of treatment before irreversible damage has occurred. However, diagnosis is commonly delayed well into adulthood because of the nonspecific character of initial symptoms. Here, we report findings in a neonate who was evaluated because of a positive family history of OAT deficiency. The reversed enzymatic flux in early infancy resulted in borderline low ornithine concentration – evoking urea cycle disturbances – and increased proline. In addition, plasma citrulline was low. Consequently, the proline/citrulline ratio in plasma was increased compared to controls. To find out whether amino acid profiling in neonatal dried blood spots is suitable to detect OAT deficiency, we evaluated the original newborn dried blood spots of two affected patients and compared it with a database of >450,000 newborns tested in Minnesota since 2004. Proline concentrations (777 and 1,381 μmol/L) were above the 99 percentile (776 μmol/L) of the general population, and citrulline concentrations (4.5 and 4.9 μmol/L) only just above the 1 percentile (4.37 μmol/L). The proline/citrulline ratio was 172.9 and 281.8, respectively. This ratio was calculated retrospectively in the normal population, and the 99 percentile was 97.6. Applying this ratio for NBS could lead to early and specific detection of neonatal OAT deficiency, with no additional expense to newborn screening laboratories quantifying amino acids. Given that early diagnosis of OAT disease can lead to earlier treatment and prevent visual impairment, further studies are indicated to evaluate whether newborn screening for OAT deficiency is warranted.
Introduction
Ornithine-δ-aminotransferase (OAT) deficiency (OMIM: 258870) is a rare congenital metabolic disorder characterized by gyrate atrophy (GA) of the choroid and retina (Simell and Takki 1973). OAT is a nuclear-encoded pyridoxal phosphate requiring enzyme that catalyzes the conversion of excess ornithine – generated from arginine in the urea cycle – into delta pyrroline-5-carboxylic acid (P5C). Patients with GA have 10–20-fold elevations in plasma ornithine concentration and overflow ornithinuria (Simell and Takki 1973; Takki 1974). As ornithine is a precursor of proline, a deficiency in OAT leads to decreased formation of proline (Fig. 1).
Fig. 1.
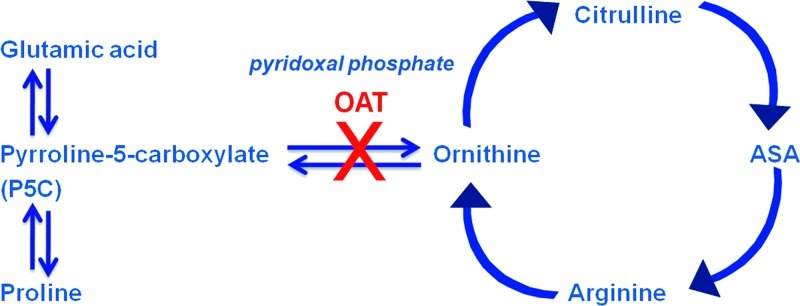
Ornithine metabolic pathway. OAT ornithine aminotransferase, ASA argininosuccinic acid
Most patients affected with OAT deficiency present in childhood with myopia followed by the development of night blindness, cataracts, and progressive constriction of vision fields, leading to blindness in the fourth to fifth decade (Kaiser-Kupfer et al. 1985; Takki and Milton 1981). In addition, a high prevalence of neurological impairment has been reported in patients with OAT deficiency (Valayannopoulos et al. 2009). The pathophysiology of the chorioretinal degeneration in GA is not fully understood. Putative pathogenic factors include proline deficiency in the choroid and retina (Hayasaka et al. 1985), elevated intraocular concentrations of ornithine (Kaneko et al. 2007), and catabolic products of ornithine (Sulochana et al. 2000).
It has been argued that treatment of OAT deficiency is not useful in clinically diagnosed patients since treatment has no effect on the function and histology of the visual system. Delineation of the efficacy of this treatment is hampered by the symptomatic phase at which diagnosis is usually made and the irreversible ocular damage that is already present at time of diagnosis. Notably, an arginine-restricted diet (arginine being the immediate precursor of ornithine; Fig. 1) has been shown to reduce ornithine levels and prevent retinal degeneration in an OAT−/− mice. Since it was reported that the age at start of treatment was the most important factor supporting the ability to comply with diet (Santos et al. 2006) and since OAT deficiency is associated with a vitamin B6-responsive phenotype (Ohkubo et al. 2005), a case can be made in favor of early diagnosis.
We and others have diagnosed cases with OAT in infancy (Cleary et al. 2005; Champion et al. 2002; Webster et al. 1999), before ocular damage had occurred. Newborns with OAT deficiency may develop hyperammonemia which may lead to poor feeding, vomiting, seizures, or coma. The hyperammonemia can be explained by the reversed flux of the OAT reaction in early life, in the direction of ornithine synthesis rather than ornithine degradation (Fig. 2). Consequently, OAT deficiency causes a relative deficiency of ornithine and its products arginine and citrulline, causing an impairment of the urea cycle functioning (hence the hyperammonemia). Based on these observations, we hypothesized that indices of a block of the reversed enzymatic flux could be used as a tool for neonatal diagnosis of this disease, including the analysis of dried blood spots routinely collected for newborn screening.
Fig. 2.
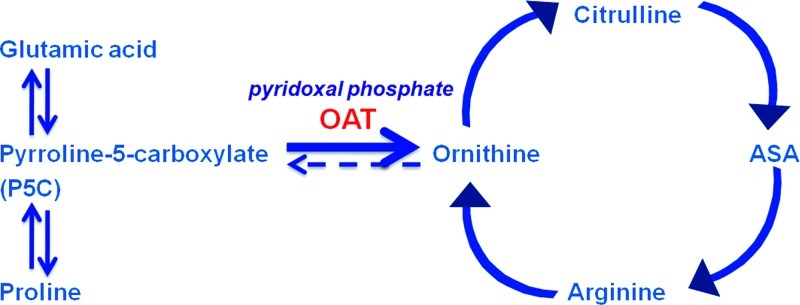
Ornithine metabolic pathway in the neonate. OAT ornithine aminotransferase, ASA argininosuccinic acid
Case Reports
Case 1 is the fourth child of consanguineous Moroccan parents. His eldest sister (case 2) was diagnosed with OAT deficiency after presenting in infancy with symptomatic hyperammonemia (Cleary et al. 2005). In case 1, evaluation 17 days after birth was scheduled in an attempt to either rule out or confirm the diagnosis presymptomatically. Frequent crying and lack of feeding rhythm were noted, but no other abnormalities. Blood ammonia concentration at that time was 101 μmol/L. Physical examination was uneventful. Amino acid analysis was performed (Table 1). Four weeks later (age 38 days), parents reported repeated vomiting at night after feeding. He was reported to look ill, foul, and drowsy after vomiting. Physical examination revealed an alert infant with a mild rash, but no other abnormalities, particularly no organomegaly. Biochemical investigation at this time showed elevated ammonia (230 μmol/L, n: <50 μmol/L). Hyperammonemia corrected rapidly after introducing a protein-restricted diet and arginine supplementation.
Table 1.
Plasma amino acids concentrations in two untreated patients with OAT deficiency
| Glutamine (μmol/L) | Proline (μmol/L) | Citrulline (μmol/L) | Ornithine (μmol/L) | Arginine (μmol/L) | Proline/citrulline | |
|---|---|---|---|---|---|---|
| Normal | 402–776 | 97–254 | 8–36 | 41–129 | 7–128 | <32 |
| Case 1 | ||||||
| 17 days | 877 | 1,090 | 4 | 56 | 14 | 272 |
| 38 days | 1,228 | 1,089 | <1 | 32 | 12 | >1,089 |
| Case 2 | ||||||
| 71 days | 570 | 3 | 35 | 42 | 190 | |
Case 3 is a male who presented at 8 weeks with respiratory difficulties, encephalopathy, a respiratory alkalosis, and hyperammonemia (355 μmol/L). Treatment with arginine, benzoate, and phenylbutyrate resulted in rapid recovery. OAT enzyme activity in fibroblasts was undetectable (control mean 0.40 ± 0.09 (n = 5) μmol pyrroline-5-carboxylate formed per mg protein per hour).
Material and Methods
Amino acids in plasma and urine were analyzed by means of automated ion-exchange chromatography with postcolumn ninhydrin derivatization. Plasma amino acid analyses were performed on a JEOL AminoTac and urine on a Biochrom 30. Orotic acid was analyzed using selected-reaction monitoring (SRM) analysis on a Micromass Quattro LC triple quadrupole mass spectrometer (LC-MS/MS).
Blood spot from case 1 was collected as part of the neonatal screening for inborn errors of metabolism in the Netherlands, 3 days after birth. Blood spot from case 3 was collected as part of Leeds neonatal screening, 6 days after birth. The patients’ original NBS cards were obtained after informed parental consent. No information is given about the storage conditions of the blood spot from case 3.
Ten control blood spot samples were obtained from the same sampling day as case 1 to exclude differences in storage conditions. Control blood spot samples were provided by the RIVM (Bilthoven) after removal of all personal identifying information. All blood spots were reevaluated for amino acid profile at the same time (Turgeon et al. 2008). These data were compared to >450,000 blood spot samples analyzed as part of Minnesota NBS program. The determination of amino acids was performed according to a routine procedure for amino acid analysis in DBS with LC-MS/MS.
Results
In urea cycle defects, ammonia and its precursors (e.g., glutamine) accumulate. Analysis of urea cycle intermediates showed a normal concentration of plasma ornithine and arginine in the patient while plasma citrulline was decreased (Table 1), an abnormal biochemical profile that was also reported in the sibling of this patient (Cleary et al. 2005; Table 1: case 2). Plasma concentrations of arginine and ornithine were normal, suggesting that the marginal ammonium detoxification capability during the early days of life is sufficient to maintain normal levels of ornithine and arginine. Interestingly, plasma proline was strongly increased. Retrospective analyses of plasma and urine of the sibling (on the age of 71 days; Table 1) revealed increased proline concentration in plasma as well (Table 1). Orotic aciduria and an increased urine proline excretion were observed in both patients (Table 2). Biochemical analysis of case 3 was performed in Leeds at 57 days of age after the commencement of arginine therapy. This revealed a raised plasma ornithine (632 μmol/L, ref 58–251) and low proline (90 μmol/L, ref 110–322). The other amino acids in plasma were normal. In addition, orotic aciduria was not present.
Table 2.
Urine biochemical characteristics in two untreated patients with OAT deficiency
| Orotic acid (μmol/mmol creatinine) |
Proline (μmol/mmol creatinine) |
|
|---|---|---|
| Normal | <3 | 21–213 (<4 weeks) 0–130 (1–6 months) |
| Case 1 | ||
| 17 days | 22 | 3,692 |
| Case 2 | ||
| 58 days | 39 | 320 |
Retrospective analysis of the original NBS sample of case 1 and case 3 revealed a proline concentration of 777 and 1,381 μmol/L, above the 99 percentile (737 μmol/L) of the 10 NBS analyzed. This is in accordance when compared with the 99 percentile (776 μmol/L) of >450,000 cases from the Minnesota database. Citrulline (4.5 and 4.9 μmol/L) was just below and equal to the 1 percentile (4.9 μmol) of the ten Dutch controls and just above the 1 percentile (4.37 μmol) of the Minnesota historical data.
An increased ratio of proline/citrulline (172.9 and 281.8) was found compared to the ten Dutch controls with a 99 percentile of 83.6 (Fig. 3, dashed line). The ratio was then applied retrospectively to NBS samples (>450,000) submitted for routine NBS and estimated to be 97.6 (Fig. 3, solid line).
Fig. 3.

Proline/citrulline ratio and proline concentration in the neonatal dried blood spot of two affected patients with OAT deficiency (case 1 and case 3). Pro proline, Cit citrulline. The dashed line indicates the 99 percentile of control samples (83.6) obtained from the same sampling day as case 1. Diamond indicates the individual control samples. The solid line indicates the 99 percentile (97.6) of >450,000 blood spot samples analyzed as part of Minnesota NBS program
Discussion
This study has shown that newborn screening for OAT deficiency may be feasible by determination of the proline/citrulline ratio. Although only two cases are reported, there is a clear disease marker ratio (Fig. 3), and data have been compared with >450,000 control subjects. Ornithine concentration at birth (case 1) was within normal limits. This is in accordance with our previous study (Cleary et al. 2005) and with the study of Wang et al. who showed that ornithine rises to abnormally high levels at 3–4 months after birth (Wang et al. 1996), well past the time that the newborn screening blood spot is collected. The increased proline/citrulline ratio also supports the hypothesis that the net flux in the OAT reaction in neonates is in the direction of ornithine synthesis rather than ornithine degradation.
OAT deficiency is a genetic condition that involves the choroid and retina. Since it has been hypothesized that direct ornithine toxicity to retinal cells, particularly to the RPE (retinal pigment epithelium), is involved in the earliest stages of the pathophysiology of OAT deficiency and since ornithine is not increased in the neonatal period, this unique feature of nature (reversibility of the enzyme) gives us a broad time window for effective therapeutical intervention. It is plausible that screening for OAT deficiency and early treatment may not only prevent irreversible damage of the eyes but may also prevent neurological impairment as hyperammonemia can be avoided. Insight in the natural history of this disease may help to further develop therapeutical trials.
Because there is effective treatment for patients with an OAT deficiency (Kaiser-Kupfer et al. 2004; Weleber and Kennaway 1981), screening for this disease fully meets the recommendations by the American College of Medical Genetics (Watson et al. 2006). In addition, OAT deficiency has been found in several ethnic groups around the world with a particularly high incidence in Finland with an estimated frequency of about 1 in 50,000 (Heinanen et al. 1998). This makes OAT deficiency, at least in Finland, a more common disorder than many of the diseases currently screened for. The implementation of newborn screening for OAT deficiency would not require new technologies and/or markers since proline and citrulline are already included in several newborn screening programs in the United States.
The differential diagnosis of neonatal hyperammonemia should include OAT deficiency. Biochemical diagnosis may be challenging since hyperornithinemia may be absent (case 1). However, the proline/citrulline ratio in plasma of our patient was strongly increased. In addition, retrospective plasma analyses of the sibling with proven OAT deficiency (at the age of 71 days) also revealed an increased proline/citrulline ratio. Currently, there are ten patients with OAT deficiency known in the Netherlands [data provided by the Dutch Diagnosis Registration Metabolic Diseases (DDRMD)]. The vast majority of these cases were diagnosed after childhood (>16 years of age). The rarity of the neonatal OAT presentation and/or the failure to correctly diagnose young patients limits the number of patients available to study. Reports with proven neonatal OAT deficiency (Champion et al. 2002; Webster et al. 1999) are scarce and do not report on proline concentrations in plasma. From this study, we suggest that calculating proline/citrulline ratio in plasma may help to adequately diagnose OAT deficiency in the neonatal period especially since enzyme analysis is cumbersome and over 50 different mutations (most point mutations) have been reported (Brody et al. 1992).
It is unknown at this time how reliably the minority of patients (<5%) which are responsive to therapy with vitamin B6 could be detected when screening for OAT deficiency.
The present finding shows that gyrate atrophy due to OAT deficiency may be one of the few hereditary degenerative retinopathies, which may be prevented if this disorder is included in newborn screening and that such screening is likely to be feasible, practical, and ethically acceptable.
Acknowledgments
We like to thank David McHugh for technical support on the MN database and Dr. Gepke Visser for support on the DDRMD (Dutch Diagnosis Registration Metabolic Diseases).
Take Home Message
Determining the proline/citrulline ratio in plasma as well as in dried blood spots routinely collected for newborn screening is a tool for neonatal diagnosis of OAT deficiency.
Footnotes
Competing interests: None declared
References
- Brody LC, Mitchell GA, Obie C, et al. Ornithine delta-aminotransferase mutations in gyrate atrophy. Allelic heterogeneity and functional consequences. J Biol Chem. 1992;267:3302–3307. [PubMed] [Google Scholar]
- Champion MP, Bird S, Fensom T, Dalton RN. Ornithine aminotransferase deficiency (gyrate atrophy) persenting with hyperammonaemic encephalopathy. J Inherit Metab Dis. 2002;25(supplement 1):29. [Google Scholar]
- Cleary MA, Dorland L, de Koning TJ, et al. Ornithine aminotransferase deficiency: diagnostic difficulties in neonatal presentation. J Inherit Metab Dis. 2005;28:673–679. doi: 10.1007/s10545-005-0074-1. [DOI] [PubMed] [Google Scholar]
- Hayasaka S, Saito T, Nakajima H, Takahashi O, Mizuno K, Tada K. Clinical trials of vitamin B6 and proline supplementation for gyrate atrophy of the choroid and retina. Br J Ophthalmol. 1985;69:283–290. doi: 10.1136/bjo.69.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinanen K, Nanto-Salonen K, Leino L, et al. Gyrate atrophy of the choroid and retina: lymphocyte ornithine-delta-aminotransferase activity in different mutations and carriers. Pediatr Res. 1998;44:381–385. doi: 10.1203/00006450-199809000-00019. [DOI] [PubMed] [Google Scholar]
- Kaiser-Kupfer MI, Ludwig IH, de Monasterio FM, Valle D, Krieger I. Gyrate atrophy of the choroid and retina. Early findings. Ophthalmology. 1985;92:394–401. doi: 10.1016/s0161-6420(85)34022-8. [DOI] [PubMed] [Google Scholar]
- Kaiser-Kupfer MI, Caruso RC, Valle D, Reed GF. Use of an arginine restricted diet to slow progression of visual loss in patients with gyrate atrophy. Arch Ophthalmol. 2004;122:982–984. doi: 10.1001/archopht.122.7.982. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Ando A, Okuda-Ashitaka E, et al. Ornithine transport via cationic amino acid transporter-1 is involved in ornithine cytotoxicity in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2007;48:464–471. doi: 10.1167/iovs.06-0398. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Ueta A, Ito T, et al. Vitamin B6-responsive ornithine aminotransferase deficiency with a novel mutation G237D. Tohoku J Exp Med. 2005;205:335–342. doi: 10.1620/tjem.205.335. [DOI] [PubMed] [Google Scholar]
- Santos L, Fiona WJ, Walter JH. Dietary compliance in ornithine aminotransferase deficiency. J Inherit Metab Dis. 2006;29:240. doi: 10.1007/s10545-006-0286-z. [DOI] [PubMed] [Google Scholar]
- Simell O, Takki K. Raised plasma-ornithine and gyrate atrophy of the choroid and retina. Lancet. 1973;1:1031–1033. doi: 10.1016/S0140-6736(73)90667-3. [DOI] [PubMed] [Google Scholar]
- Sulochana KN, Ramakrishnan S, Mahesh L, Punitham R. Possible role of polyamines in gyrate atrophy. Indian J Ophthalmol. 2000;48:37–43. [PubMed] [Google Scholar]
- Takki K. Gyrate atrophy of the choroid and retina associated with hyperornithinaemia. Br J Ophthalmol. 1974;58:3–23. doi: 10.1136/bjo.58.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takki KK, Milton RC. The natural history of gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88:292–301. doi: 10.1016/s0161-6420(81)35031-3. [DOI] [PubMed] [Google Scholar]
- Turgeon C, Magera MJ, Allard P, et al. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem. 2008;54:657–664. doi: 10.1373/clinchem.2007.101949. [DOI] [PubMed] [Google Scholar]
- Valayannopoulos V, Boddaert N, Mention K, et al. Secondary creatine deficiency in ornithine delta-aminotransferase deficiency. Mol Genet Metab. 2009;97:109–113. doi: 10.1016/j.ymgme.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Wang T, Milam AH, Steel G, Valle D. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J Clin Invest. 1996;97:2753–2762. doi: 10.1172/JCI118730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P, Howell RR. Newborn screening:toward a uniform screening panel and systeom executive summary. Genet Med. 2006;8:1S–11S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster M, Allen J, Rawlinson D, Brown A, Olpin S, Leonard JV. Ornithine aminotransferase deficiency presenting with hyperammonaemia in a premature newborn. J Inherit Metab Dis. 1999;22(supplement 1):80. [Google Scholar]
- Weleber RG, Kennaway NG. Clinical trial of vitamin B6 for gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88:316–324. doi: 10.1016/s0161-6420(81)35035-0. [DOI] [PubMed] [Google Scholar]