

Abstract
The bacterial flagellar motor is an intricate nanomachine powered by a transmembrane electrochemical gradient. Rotation is driven by the cumulative action of several peptidoglycan-anchored stator complexes on the rotor. In proton-motive force-driven motors, the stator complex is composed of a motility protein B (MotB) dimer surrounded by four copies of MotA, where both MotA and MotB are integral membrane proteins. The lack of full-length MotA and MotB structures hinders understanding of the mechanism of torque generation. Given the low levels of expression and low stability of detergent-solubilized MotB, a soluble chimaeric variant was engineered, where the two transmembrane helices of the MotB dimer were replaced by a leucine zipper. The biochemical and biophysical analysis of the resultant protein showed that it was properly folded, stable, behaved as a monodisperse dimer at low pH, had molecular dimensions close to those expected for native MotB and yielded reproducible crystals. The chimaeric protein is, therefore, a good candidate for structural studies. This ‘solubilization by design’ approach may be generally applicable to the production of soluble forms of other dimeric, trimeric and tetrameric single-span membrane proteins for functional and structural studies.
Keywords: flagellar motor, membrane proteins, motility protein B, protein engineering
1. Introduction
Motility protein B (MotB) is a key component of the energy-coupling stator complex that drives rotation of the bacterial flagellum, providing motility and enhancing pathogenicity of many bacteria [1–5]. The stator is part of the membrane-embedded basal body of the flagellum. The basal body is a rotary motor driven by the ion-motive force. The core central structure of its rotor (the rod) is attached to a long helical filament via a hook [5,6]. The rod is surrounded by several ring structures (MS-ring, C-ring and, in Gram-negative bacteria, additional peptidoglycan (PG)-associated L- and P-rings) [5,7]. Rotation is driven by the cumulative action of several circumferentially positioned PG-anchored stator complexes on the C-ring [8].
The stator complex of the proton-motive force-driven motor is composed of proteins MotA and MotB, and functions as a proton conduction channel. In this complex, a central MotB dimer surrounded by four copies of MotA (figure 1) [9–11]. Each stator complex functions independently to provide a pathway for protons across the cytoplasmic membrane and generate torque [5,8,10,12]. The mechanism by which the proton-motive force is coupled with torque generation is, as yet, unknown.
Figure 1.
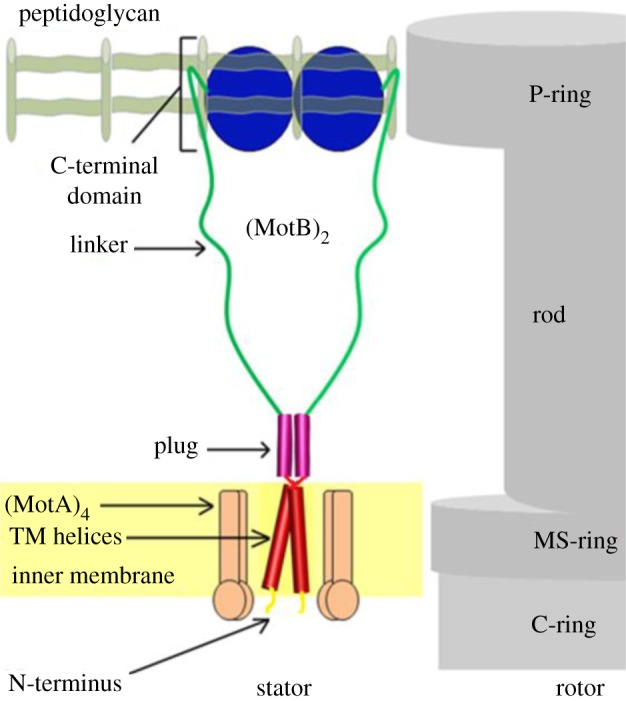
Schematic of the stator and rotor components. In the stator complex, the MotB forms a dimer, each half of which is anchored to the inner membrane via its N-terminal TM helix (drawn in red), and to PG via its C-terminal domain (blue). (Online version in colour.)
In the stator unit incorporated into the motor, each MotB monomer is anchored at one end to the cytoplasmic membrane via a single N-terminal hydrophobic α-helix, and at the other end to PG via its C-terminal globular domain (figure 1). Analysis of the crystal structures of the PG-binding domain of MotB from Helicobacter pylori [13–15] and Salmonella typhimurium [16] elucidated the mechanism by which the 70 Å wide C-terminal part of the MotB dimer can insert into the pores of the PG matrix, the smallest diameter of which was estimated to be 70 Å [17]. A C-terminal periplasmic extension of the transmembrane (TM) helix (residues 52–65 in Escherichia coli MotB, 41–54 in H. pylori MotB) is predicted to fold into an amphipathic helix, termed ‘plug’ (figures 1 and 2a). The ‘plug’ helix and C-terminal PG-binding domain are connected by a linker. The ‘plug’ helix is believed to play an essential role in suppressing premature proton flow-through the stator complex until the latter incorporates into the motor [19]. Dimerization of MotB within the stator occurs via the PG-binding domain, ‘plug’ helix and TM helix, and is known to be important for function (figure 1) [13,16,19,20].
Figure 2.

The chimaera design showing the amino acid sequence, predicted secondary structure and elements for which the three-dimensional structure is known. (a) Alignment of the TM and the ‘plug’ region in representative MotB sequences. The sequences are shown for H. pylori 26695 (Hp; UniProt P56427), Rhodobacter sphaeroides WS8 (Rs; UniProtKB/TrEMBL A3PKW2), Bacillus subtilis (Bs; UniProtKB/Swiss-Prot entry P28612), Aquifex aeolicus VF5 (Ae; SWISS-PROT/TrEMBL O67121) and Escherichia coli (Ec; UniProtKB/Swiss-Prot entry P0AF06). Conserved residues are highlighted in red. The heptad repeat pattern is shown above the sequences for the ‘plug’ helix. (b) Schematics of native MotB and the soluble MotB variants used in this study. GCN4 zipper indicates the GCN4-derived leucine zipper, PBD denotes the peptidoglycan-binding domain. (c) The sequence and structure of the GCN4 zipper and C-terminal domain of MotB (residues 125–256) (PDB RCSB: 2ZTA [18], 3CYP [13]) are boxed with orange and blue solid lines, respectively. The plug helix is boxed with a magenta dotted line. The heptad repeat pattern hpphppp is shown under the fusion site of the chimaera (h is hydrophobic and p is polar). Predicted secondary structure is shown above the sequence. (Online version in colour.)
Despite significant progress in characterization of the PG-binding domain, full-length MotB has so far resisted structure determination. Previously reported crystal structures of N-terminally truncated MotB variants [13–16] contain only part of the linker. The lack of the structural information about the linker and ‘plug’ helix in the context of full-length protein thwarts progress towards understanding of the mechanism of stator activation and torque generation. Full-length MotB is unsuitable for structural studies owing to instability when expressed on its own [21]. Our previous attempts to crystallize it yielded crystals of breakdown products [15,22]. Here, we describe the design, expression, purification, crystallization and biophysical characterization of a close structural mimic of full-length MotB—a soluble chimaeric variant of H. pylori MotB (chimMotB), in which the helical TM domain was replaced with a leucine zipper (dimerization) motif derived from the yeast transcription factor GCN4 [18]. The parallel zipper possesses a pair of intertwining helices stabilized by hydrophobic interface residues. The parallel coiled coil homodimer of the GCN4 and ‘plug’ regions in the designed chimaera closely resembles that of the TM and ‘plug’ helices in native MotB (figure 1), thus preserving the native MotB fold in the rest of the construct. We found that the approach described here produced large quantities of soluble, stable, properly folded protein that behaved as a monodisperse dimer and had molecular dimensions close to those expected for native MotB. This ‘solubilization by design’ approach may be generally applicable to the production of soluble forms of other dimeric, trimeric and tetrameric single-span membrane proteins for functional and structural studies.
2. Material and methods
2.1. Construction of the recombinant plasmids and overexpression
The plasmid for expression of H. pylori MotB-C97 (C-terminal fragment 97–256 comprising the PG-binding domain and part of the linker; figure 2b) was previously described [15]. The coding sequence for MotB-C40 (residues 40–256 comprising the PG-binding domain, linker and ‘plug’ figure 2b) was PCR-amplified from genomic DNA of strain 26695 of H. pylori using KOD Hot Start DNA polymerase (Novagen) and the primers CACCAACAAATCCAAAGTGGAAGCCTTAAAAAC (forward) and TCATTCTTGCTGTTTGTGCGGATTG (reverse). The amplified fragment was cloned into the pET151/D-TOPO vector using the TOPO cloning kit (Invitrogen) to produce the expression vector that contains an N-terminal His6-tag followed by a TEV protease-cleavage site. The expression clone was confirmed by DNA sequencing. The vector was transformed into E. coli strain BL21-CodonPlus (DE3)-RIPL (Stratagene). Cells were grown in Luria–Bertani (LB) medium containing 100 mg l–1 ampicillin and 34 mg l−1 chloramphenicol at 37°C until an optical density at 600 nm (OD600) of 0.6 was reached, at which point overexpression of MotB-C40 was induced by adding 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and growth continued for a further 3 h. The cells were then harvested by centrifugation at 6000g for 20 min at 4°C.
The amino acid sequence of chimMotB contains the N-terminal GCN4 leucine zipper peptide MKQLEDKVEELLSKNYHLENEVARL (Genbank AAA34640.1, residues 250–274) fused to the C-terminal periplasmic domain of H. pylori MotB (residues 41–256, gene HP0816, H. pylori 26695 genome [23] (figure 2). The gene encoding chimMotB was synthesized by GenScript (USA) and inserted into the pET151/D-TOPO vector. The vector was then transformed into E. coli strain Rosetta 2 (Novagen). Cells were grown in LB medium containing 50 mg l−1 ampicillin and 34 mg l−1 chloramphenicol at 37°C until an OD600 of 0.8 was reached, at which point overexpression of chimMotB was induced by adding 0.5 mM IPTG and growth continued for a further 3 h, after which the cells were harvested.
2.2. Protein purification
MotB-C97 was purified by following the previously published procedure [15,22]. Cells expressing chimMotB or MotB-C40 were lysed using a freeze–thaw method followed by sonication in buffer A (20 mM Tris/HCl pH 7.4, 200 mM NaCl and 1 mM PMSF) and three passages through an Avestin cell disruptor. Cell debris was removed by centrifugation at 10 000g for 20 min, after which the cell lysate was cleared by further centrifugation at 34 000g for 40 min to remove inclusion bodies, aggregates and membranes. NaCl and imidazole were then added to the supernatant to the final concentrations of 500 and 15 mM, respectively, after which the supernatant was loaded onto a 5 ml Hi-Trap Chelating HP column (GE Healthcare) pre-washed with buffer B (20 mM Tris/HCl pH 7.4, 500 mM NaCl, 15 mM imidazole, 1 mM PMSF). The column was washed with 20 column volumes of buffer C (20 mM Tris/HCl pH 7.4, 500 mM NaCl, 80 mM imidazole, 1 mM PMSF). To remove E. coli DnaK contamination, the column was then subjected to 10 rounds of incubation in 2.5 ml of buffer B containing 2 mM MgATP for 10 min and a wash with buffer C. Protein was eluted with buffer C containing 500 mM imidazole. The N-terminal tag was cleaved off with His6-TEV protease (Invitrogen), while dialysing the sample overnight at 4°C against buffer D (50 mM Tris/HCl pH 8.0, 0.5 mM EDTA, 2 mM DTT, 200 mM NaCl, 1% glycerol). NaCl and imidazole were then added to the sample to the final concentrations of 500 and 15 mM, respectively, and the TEV protease and uncleaved protein were removed over a Hi-Trap Chelating HP column. The flow-through was concentrated to 15 ml in a VivaSpin 10 000 Da cut-off concentrator, exchanged into buffer E (30 mM Tris/HCl pH 7.4) by passing through a HiPrep 26/10 Desalting column (GE Healthcare), and loaded onto a Resource Q column (GE Healthcare) pre-equilibrated in buffer E containing 50 mM NaCl. chimMotB was eluted with a linear gradient of 50–600 mM NaCl in buffer E, concentrated to 4 ml and loaded onto a Superdex 200 HiLoad 26/60 gel-filtration column (GE Healthcare) pre-equilibrated with buffer F (100 mM sodium acetate pH 4.6 and 200 mM NaCl).
2.3. Circular dichroism spectroscopy
MotB-C40, MotB-C97 and chimMotB were dialysed exhaustively against buffer F. Far-UV circular dichroism (CD) spectra were recorded at a protein concentration of 0.2 mg ml−1 at 20°C using a JASCO J600 spectrapolarimeter (calibrated with 0.06% d-10 camphorsulphonic acid) over the wavelength range 190–260 nm with the scan rate of 10 nm min−1. Spectra were recorded in triplicate and averaged. The percentage of secondary structure was calculated by deconvoluting the CD spectra using the program K2d from the DichroWeb CD secondary structure server (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml; [24]).
Thermal denaturation measurements were performed in a 0.2 cm path-length thermostated quartz cell at a protein concentration of 0.2 and 0.1 mg ml−1 for MotB-C40 and chimMotB, respectively, in buffer F using a Jasco J-815 CD spectropolarimeter. A heating rate of 60°C h−1 was applied, and the CD signal at 222 nm was monitored. The thermal denaturation data were fit to a derivation of the Boltzmann equation for the two-state unfolding model using Graph Pad Prism 5 to obtain the midpoint of denaturation (the thermal melting point) [25].
2.4. Matrix-assisted laser desorption ionization time of flight mass spectrometry
One microlitre of the purified chimMotB was mixed with 1 μl of α-cyano-4-hydroxycinnamic acid matrix solution, placed on the matrix-assisted laser desorption ionization sample plate and allowed to dry. Molecular mass was analysed by an Applied Biosystems 4700 Proteomics Analyser.
2.5. Size-exclusion chromatography and multiangle laser light scattering/quasi-elastic light scattering analysis
Purified chimMotB was dialysed exhaustively against buffer F and concentrated to 2 mg ml−1. A 50 µl sample was loaded onto a Superdex 200 5/150 gel-filtration column (GE Healthcare), equilibrated with buffer F flowing at 0.2 ml min−1. The eluate was passed through an in-line DAWN HELEOS II laser photometer (λ = 658 nm), an Optilab T-rEX differential refractive index detector (Wyatt Technologies) and a dynamic light scattering detector (WyattQELS, Wyatt Technologies). A bovine serum albumin (BSA) standard was run to normalize the multiangle laser light scattering (MALLS) detectors. Data were analysed in ASTRA 6.0 (Wyatt Technologies), with a value for the refractive index increment (dn/dc)protein of 0.185 ml g−1. The results are presented in table 1.
Table 1.
Molecular weights determined by SEC MALLS/QELS analysis.
| experimental MW (kDa) | theoretical MW (kDa) | polydispersity | |
|---|---|---|---|
| BSA | 65.3±1.2 | 66.5 | 1.001±0.7% |
| chimMotB | 55.9±1.4 | 56.1 (dimer) | 1.000±0.2% |
2.6. Small-angle scattering
Room-temperature small-angle X-ray scattering (SAXS) data were acquired for a 0.250 mg ml−1 chimMotB solution in buffer F placed in a 1.5 mm quartz capillary using the SAXS beamline at the Australian Synchrotron equipped with a Pilatus Detector. To collect scattering data, chimMotB and a matching buffer solution were exposed to X-rays for 1 s as the sample flowed through the capillary. The resultant two-dimensional scattering images were radially averaged and normalized for sample transmission. After scaling, scattering intensities of the buffer and empty capillary were subtracted from the intensities of the chimMotB solution. Data analysis was performed using the ATSAS suite of software [26]. The program Primus [27] was used to calculate a Guinier plot to determine values for zero-angle scattering I(0) and radius of gyration (Rg), using a 0.8 cut-off for sRg, where s is the magnitude of the scattering vector. The pair distribution function P(r) was calculated by the program GNOM [28] as an indirect Fourier transform of the scattering curve I(s). This function provides the probabilities of distances between the scattering centres and the maximum dimension of the scattering molecular species [28]. The maximum dimension of chimMotB (Dmax) was calculated by restraining the P(r) function to 0 at rmax and varying rmax over a wide range to find the value (rmax = Dmax) that gave a plausible P(r) function and yielded the highest ‘total estimate’ value. Additionally, I(0) was computed using GNOM. The I(0) and Rg values calculated from the Guinier approximation or the P(r) were comparable. To estimate the molecular mass of chimMotB, the forward scattering I(0) of chimMotB was determined on the absolute scale using the known scattering of water as previously described [29]; the partial specific volume of the protein was assumed to be 0.74 cm3 g−1. Theoretical calculations of the hydrodynamic radius from the crystal structure were carried out as described by Ortega et al. [30] using HYDROPRO v. 10 (http://leonardo.inf.um.es/macromol/programs/hydropro/hydropro.htm).
2.7. Crystallization
The chimMotB protein was dialysed exhaustively against buffer G (30 mM sodium acetate, pH 4.6), concentrated to 13 mg ml−1 and centrifuged for 40 min at 34 000g to clarify the solution. Screening of crystallization conditions was carried out by the sitting-drop vapour-diffusion method at 20°C using an automated crystallization robot Phoenix (Art Robbins Instruments). The crystallization droplets contained 100 nl of the protein solution mixed with 100 nl of the reservoir solution and equilibrated against 100 μl of the reservoir solution in a 96-well Intelliplate (Art Robbins Instruments). Rod-like crystals of dimensions up 0.02 × 0.02 × 0.1 mm appeared after two weeks from condition 85 of the Crystal Screen HT (Hampton Research) containing 10% (w/v) PEG 8000, 8% (w/v) ethylene glycol and 100 mM HEPES/NaOH pH 7.5. In addition, hexahedron crystals of dimensions 0.05 × 0.05 × 0.05 mm grew after three weeks from condition 55 of the PEG/Ion Screen (Hampton Research) containing 12% (w/v) PEG 3350 and 100 mM sodium malonate pH 7.0.
3. Results
3.1. Design of soluble MotB chimaera
The TM helices of the two native MotB molecules associate into a parallel symmetric dimer that structurally resembles a coiled coil (figure 1) [20]. It has previously been postulated that in the activated stator complex, the ‘plug’ helices just C-terminal to the TM helices associate with each other via their hydrophobic faces into a parallel coiled coil that forms an extension of the TM-coiled coil (figure 1, [19]). The primary sequence of the ‘plug’ helix conforms to a heptad repeat pattern typical of soluble α-helical coiled coils (hpphppp, where h is hydrophobic and p is polar) (figure 2a). We fused the GCN4 dimerization domain in register with the ‘plug’ helix, while maintaining the heptad repeat pattern (figure 2c). In the dimer of this chimaera, the region comprising the GCN4 motif and ‘plug’ helix is predicted to form one continuous coiled coil. The parallel coiled coil homodimerization mode of the GCN4 and ‘plug’ regions in this protein is predicted to closely resemble that of the TM and ‘plug’ helices in native MotB (figure 1), thus conveying stability to the entire construct and preserving the native fold. In addition, we designed a truncated soluble MotB variant MotB-C40 that lacks the TM helix, but retains the ‘plug’ region. To facilitate the protein purification via affinity chromatography, the genes encoding chimMotB and MotB-C40 were cloned with the cleavable N-terminal polyhistidine tag into the pET151/D-TOPO vector. Both removal of the TM helix and its substitution with the GCN4 peptide greatly improved expression levels in comparison with the wild-type MotB (data not shown).
3.2. Protein purification and removal of contaminating DnaK
During purification, chimMotB and MotB-C40 co-eluted from the Ni–NTA column with contaminating approximately 65 kDa protein (figure 3; data for MotB-C40 not shown). Mass spectroscopy of the tryptic digest of the gel band identified this protein as E. coli DnaK. Analysis of the H. pylori MotB sequence (figure 2c) localized the putative DnaK binding site to a stretch of hydrophobic residues flanked by charged residues, QPVVVIPPD (residues 65–73). This is part of the MotB linker region (55–112) that is believed to be more flexible than the rest of the structure. To circumvent the unwanted contamination of DnaK, the protein bound to the purification resin was extensively washed with MgATP before elution, which dramatically reduced the amount of DnaK in the eluate (figure 3).
Figure 3.
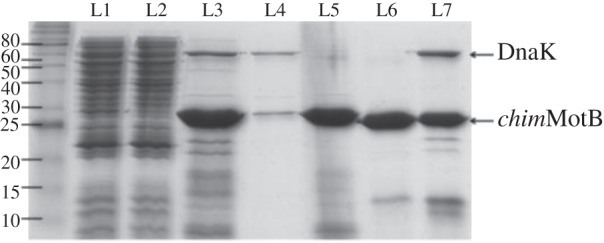
SDS–PAGE analysis of chimMotB purification. L1: total cell lysate, soluble fraction; L2: flow-through, Ni–NTA; L3: eluate during 80 mM imidazole wash, Ni–NTA; L4 eluate during 2 mM MgATP wash, Ni–NTA; L5 Ni–NTA eluate; L6: pooled peak fractions from gel-filtration step; L7: omission of the 2 mM MgATP washing step resulted in a higher level of DnaK contamination.
CD was used to investigate the secondary structure of chimMotB and compare it to that of a control protein, the MotB fragment of a known structure (MotB-C97) (figure 4). Estimation of the α-helix and β-sheet content in the secondary structure using the K2d server [24] gave the values that are close to those predicted from the sequence analysis using SOPMA [31] (table 2), indicating that purified chimMotB was folded.
Figure 4.
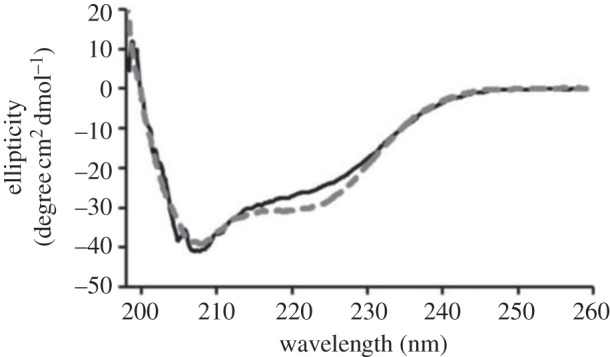
Circular dichroism (CD) spectra of MotB-C97 (solid line) and chimMotB (dashed line).
Table 2.
Secondary structure content estimated from far-UV CD spectra and predicted from the sequence analysis.
| % α-helix, % β-sheet estimation from the CD analysis | predicted % α-helix, % β-sheet | |
|---|---|---|
| MotB-C97 | 32, 17 | 33, 19 |
| chimMotB | 40, 16 | 47, 12 |
3.3. chimMotB stability against proteolysis
The time course of room-temperature proteolytic degradation of the three soluble variants of MotB, MotB-C40, MotB-C97 and chimMotB was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) analysis over a period of 6 days in 20 mM Tris pH 7.5 (figure 5). This analysis revealed that MotB-C40 undergoes slow (days rather than seconds or minutes) proteolytic cleavage by as yet unidentified contaminating proteases. SDS–PAGE and mass-spectrometry (not shown) analysis revealed that proteolysis initially yields a 22 kDa degradation product, which then further degrades to produce a stable 18 kDa fragment. In contrast, MotB-C97 and chimMotB showed no signs of proteolytic degradation over this period (figure 5). This result indicated that the addition of the N-terminal GCN4 dimerization motif to MotB-C40 (figure 2b) dramatically improves resistance of the linker region to proteolysis.
Figure 5.
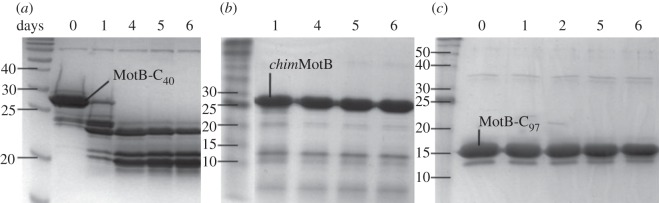
Stability of different soluble MotB variants in 20 mM Tris pH 7.5. SDS–PAGE of time-dependent degradation of (a) MotB-C40, (b) chimMotB and (c) MotB-C97 over 6 days.
3.4. chimMotB stability against thermal denaturation
We examined the thermodynamic stability of chimMotB by thermal denaturation and compared it with that of MotB-C40. The two variants displayed monophasic melting curves with very close apparent melting temperatures (66°C), indicative of similar thermodynamic stabilities (figure 6). The melting curve of chimMotB was significantly steeper than that of MotB-C40 (unfolding of chimMotB occurred over a narrow temperature range between 61°C and 70°C, whereas melting of MotB-C40 was more gradual, over the range 55°C –80°C). The highly cooperative unfolding of chimMotB suggested that it melts as a single domain, i.e. the PG-binding domain, the linker region and the N-terminal moiety, containing the GCN4 zipper and the ‘plug’ are thermodynamically coupled via intramolecular interactions. The less cooperative unfolding of MotB-C40 suggested that this variant has a less compact structure in which the interactions between the linker and PG-binding domain are likely lost.
Figure 6.
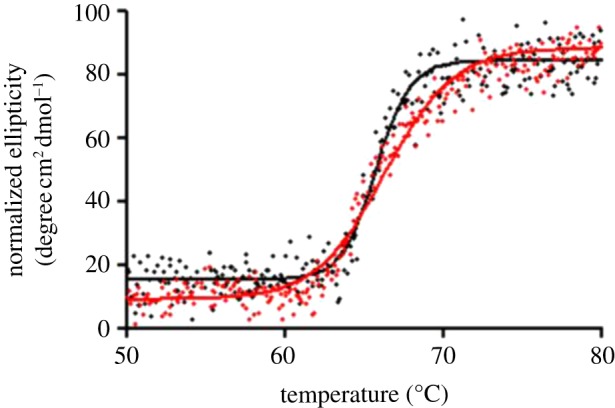
Thermal melt plots of MotB-C40 (red) and chimMotB (black). Thermal stability was measured using far-UV CD. Elipticity at 222 nm increases as chimMotB and MotB-C40 unfold.(Online version in colour.)
3.5. Monodispersity and stoichiometry of chimMotB demonstrated by light scattering analysis
To determine the oligomeric solution state, sample monodispersity and the hydrodynamic radius (Rh) of chimMotB, we carried out MALLS and quasi-elastic light scattering (QELS, also known as dynamic light scattering, DLS) analyses coupled to size-exclusion chromatography (SEC). MALLS measures the intensity of light scattered at 15 angles simultaneously for the determination of absolute molecular weights (MWs). DLS measures the time-dependent fluctuations of the light scattered by the particles to derive their hydrodynamic sizes. The elution of chimMotB from the size-exclusion column was monitored using online UV/Vis, MALLS and DLS detectors.
chimMotB eluted as a single, monodisperse peak. The derived MW value was 55.9±1.4 kDa (figure 7 and table 1; we determined the accuracy of the weight determination as approx. 2% from the quality of the BSA standard). This value is consistent with a dimer. The apparent hydrodynamic radius of the particles in this peak was 38 Å (figure 7). For comparison, calculation of the hydrodynamic radius from the crystal structure of the dimer of H. pylori MotB missing the N-terminal 63 residues (PDB RCSB 3S0Y [15]) gave the value of 37 Å.
Figure 7.

Size-exclusion chromatography and molecular weight (MW) and hydrodynamic radius determination of chimMotB. A bold solid line superimposed on the peak indicates the MW as shown on the left-hand y-axis. A dotted line represents the hydrodynamic radius Rh calculated over the central portion of the elution peak (shown by UV trace). The Rh values are shown on the right-hand y-axis.
3.6. Small-angle X-ray scattering analysis
SAXS analysis was performed on chimMotB in the same buffer as SEC MALLS. The experimental SAXS curve is displayed in figure 8a. The linearity of the Guinier plot for sRg < 0.8 (figure 8b) suggested that the sample was monodisperse and contained no aggregates. Calculation of the pair distribution function, P(r), yielded the estimate for the maximum molecular length of the particles Dmax of approximately 155 Å (figure 8c). Assuming a protein density of about 1.35 g cm−3 (average protein density), a spherical protein of the mass, equal to that of the chimMotB dimer (56 079 g mol−1) would have a diameter of 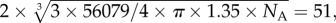 Å where NA is the Avogadro number. Our estimation of Dmax, therefore, suggested an elongated rather than spherical molecular shape for chimMotB. Indeed, the pair distance distribution function, P(r), of chimMotB showed the typical left-shifted asymmetry that is expected for elongated scattering objects (figure 8c). Estimation of the molecular mass of the scattering particles based on zero-angle scattering on an absolute scale yielded a value of 56 kDa. The estimated value is close to the theoretical molecular mass (56 kDa) of the chimMotB dimer calculated from the amino acid sequence and to the value obtained by MALLS (56 kDa), confirming that under the chosen experimental conditions chimMotB was in a dimeric state.
Å where NA is the Avogadro number. Our estimation of Dmax, therefore, suggested an elongated rather than spherical molecular shape for chimMotB. Indeed, the pair distance distribution function, P(r), of chimMotB showed the typical left-shifted asymmetry that is expected for elongated scattering objects (figure 8c). Estimation of the molecular mass of the scattering particles based on zero-angle scattering on an absolute scale yielded a value of 56 kDa. The estimated value is close to the theoretical molecular mass (56 kDa) of the chimMotB dimer calculated from the amino acid sequence and to the value obtained by MALLS (56 kDa), confirming that under the chosen experimental conditions chimMotB was in a dimeric state.
Figure 8.
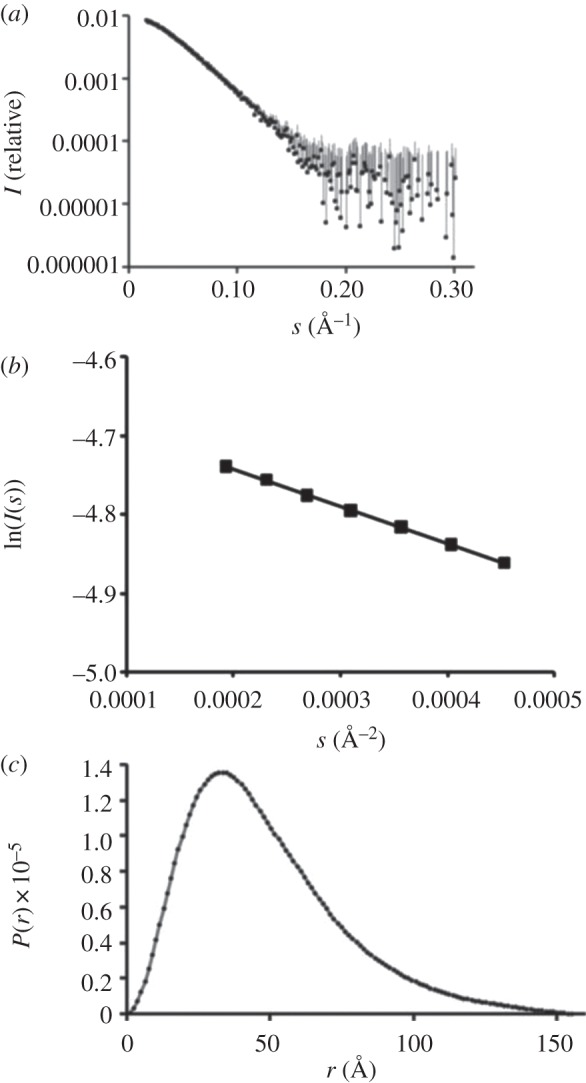
SAXS analysis of chimMotB. (a) The X-ray scattering pattern. The experimental data are displayed on logarithmic scale as dots with error bars. (b) Guinier plot within the small s range. (c) The pair distribution function showing a typical asymmetry characteristic for elongated compact molecules with a maximum dimension of approximately 155 Å.
A Guinier plot (lnI(s) versus s2, figure 8b) [32] was used to determine the radius of gyration (Rg) of the protein from the slope of the linear portion within the low s limit of the curve. The Rg value was also estimated by the GNOM program. In contrast to Guinier approximation which only uses data collected at small angles, GNOM uses data collected over all the angle range and is less sensitive to small amounts of aggregates. The two methods yielded the Rg values of 37 and 38 Å, respectively.
3.7. Preliminary crystallization
Preliminary crystals of chimMotB were obtained using high-throughput robotic screening against commercial screens containing 300 different conditions (figure 9). To assess the diffraction quality of the crystals in the high-throughput mode, the crystallization plates were placed directly in the X-ray beam of station MX2 at the Australian Synchrotron. Visual inspection of the diffraction pattern unambiguously identified these crystals as protein rather than salt. Crystal optimization and data analysis using crystallography is underway.
Figure 9.

chimMotB crystals grown using (a) 10% (w/v) PEG 8000, 8% w/v ethylene glycol and 100 mM HEPES/NaOH pH 7.5, and (b) 12% (w/v) PEG 3350 and 100 mM sodium malonate pH 7.0 as a reservoir solution, respectively. (Online version in colour.)
4. Discussion
Integral membrane proteins mediate essential functions of the cell, including energy generation, sensing and motility. Such proteins are prone to aggregation upon exogenous expression and extraction from the lipid bilayer, which makes structural and biophysical studies of them extremely challenging. Low levels of expression and low stability of these proteins in a detergent-solubilized form add to the problem. Rendering membrane proteins soluble in aqueous media by means of mutagenesis, while preserving their native-like fold, proved to be useful for boosting the expression levels and for structural and functional characterization of many important targets [33–36]. The most trivial (and most common) example of ‘solubilization by design’ involves removal of the entire TM domain [15,37]. However, the TM region in most membrane proteins plays an essential role in determining the overall fold, oligomeric state and stability, and cannot be dispensed of. This emphasizes the need for development of alternative approaches to the design of soluble variants of membrane proteins for structural and functional studies.
In this study, we have presented the rational design, purification and characterization of the water-soluble variant of homodimeric integral membrane protein MotB. The TM helices of the two native MotB molecules in the stator complex associate into a parallel symmetric dimer that resembles an α-helical coiled coil, both in the presence and absence of MotA [20]. The C-terminal extension of the TM helix, the ‘plug’, is also believed to form a parallel coiled coil and form no interactions with periplasmic loops of MotA [19]. We, therefore, argued that the overall fold of the chimaera produced by the replacement of the hydrophobic double α-helical TM domain of MotB with a water-soluble leucine zipper in register with coiled coil heptad motif in the ‘plug’ is likely to be close to the fold of the native MotB molecule. Using this rationale, we have successfully achieved ‘solubilization by design’ of dimeric flagellar protein MotB and obtained milligram amounts of soluble, highly pure, folded, stable, monodisperse, crystallizable protein for structural studies.
The soluble MotB variant produced by replacing its double α-helical TM domain with the GCN4 leucine zipper dimer showed greater stability against proteolysis relative to the soluble variant, where the TM helix was simply removed (MotB-C40). In contrast to chimMotB which remained largely intact for the duration of the experiment (6 days), MotB-C40 degraded within days to a stable 18-kDa fragment (figure 5). We have previously observed partial proteolytic degradation of N-terminally truncated MotB fragments MotB-C78 (residues 78–256), MotB-C90 and MotB-C97 during crystallization process [15]. Analysis of the electron density maps showed that some subunits in the asymmetric unit of these crystals lacked their N-terminus as a consequence of cleavage after K96 or K102 located in loop 96–107 (KATIARKGEGSV). Cleavage at this site yields an 18 kDa C-terminal fragment. The improved stability of chimMotB when compared with MotB-C40 that degrades to produce an 18 kDa fragment, therefore, suggests that in MotB-C40, residues 96–107 of the linker are solvent-exposed, poorly ordered and accessible to cleavage by contaminating proteases, whereas in chimMotB this part of the linker is well structured or shielded from the solvent.
A number of observations indicated that the MotB chimaera is folded such as native MotB. CD analysis of chimMotB revealed that its α-helix and β-sheet content is close to that predicted from the sequence analysis. The temperature denaturation studies showed that the thermal stability of chimMotB is very similar to that of MotB-C40. Furthermore, the value of the hydrodynamic radius of chimMotB calculated from the QELS analysis (38 Å) is close to that calculated for the crystal structure of the dimer of H. pylori MotB missing the N-terminal 63 residues (37 Å). In addition, two independent analyses, SEC MALLS and SAXS, revealed that chimMotB exists as a monodisperse dimer. Native MotB also functions as a dimer, in line with the previous reports of dimers observed in the crystal structures of the C-terminal periplasmic domains of MotB from H. pylori, Salmonella and Desulfovibrio vulgaris [13,15,16 and PDB ID 3KHN]. The fact that the C-terminal region of MotB lacking the ‘plug’ and the TM helix forms a dimer [13,15,16] suggests that the dimerization of chimMotB occurs via the C-terminal PG-binding domain, ‘plug’ helix and N-terminal leucine zipper.
According to the results of the scattering analyses, the chimMotB dimer has an elongated shape with the longest dimension of approximately 155 Å. In the active stator unit, membrane-anchored MotB must span an approximately 50 Å thick lipid bilayer and an additional approximately 100 Å distance in the periplasm to reach the PG layer [16,38]. The size of chimMotB is, therefore, close to that expected for native MotB. The globular C-terminal domain of MotB is approximately 70 Å in its maximum dimension [13]. Assuming the C-terminal domain adopts a similar conformation in chimMotB, the N-terminal part of the chimaera, comprising the GCN4 leucine zipper, ‘plug’ region (residues 41–54) and linker (55–112), therefore, adds at least 155 – 70 = 85 Å to the size of the molecule. One can imagine two possible configurations for the N-terminus: (i) a separate domain connected to the C-terminal PG-binding domain by an unstructured, flexible linker, or (ii) a subdomain protruding from the C-terminal domain. Improved chimMotB stability against proteolysis when compared with MotB-C40 indicates that the linker is likely well structured in the chimaera. Indeed, the sequence analysis suggests that the linker region of H. pylori MotB contains considerable secondary structure (48%; figure 2c; compare this with a 47% secondary structure content measured for the folded globular MotB-C97; table 2). Our observation of the highly cooperative temperature unfolding of chimMotB indicated that it melts as a single cooperative unit, i.e. the PG-binding domain, linker, GCN4 zipper and ‘plug’ helix are thermodynamically coupled via intra-molecular interactions within a compact structure. In contrast, the less cooperative unfolding of MotB-C40 was indicative of a more open conformation in which the interactions between the (‘plug’ plus linker) region and PG-binding domain are likely lost. Therefore, we favour configuration (ii) in which the N-terminal region of chimMotB comprising the leucine zipper (that mimics the two MotB TM helices), the ‘plug’ and the structured linker form a ‘rigid’ protrusion from the C-terminal domain as in native H. pylori MotB.
Full-length MotB has so far resisted structure determination or comprehensive biophysical and biochemical characterization owing to its low stability in isolated, detergent-solubilized form. Its water-soluble variant, chimMotB, has a fold similar to native MotB but unlike the full-length protein, the former is stable, monodisperse and forms crystals. This makes it a good candidate for structural studies by means of X-rays crystallography and other methods including SAXS, fluorescence resonance energy transfer and electron plasmon resonance.
The GCN4 dimerization motif used in this study had been extensively characterized [39] and found many applications, including stabilization of protein oligomers to facilitate crystallization. For example, replacement of the native coiled coil region of the proteasomal ATPase from Archaeoglobus fulgidus with the GCN4 zipper was previously shown to stabilize the entire oligomeric interface and promote crystallization [40]. The usability of the GCN4 leucine zipper motive is enhanced by the fact that mutations in the zipper sequence can controllably alter its oligomerization state from a dimer to either a trimer or tetramer [39]. Previously, replacing the trimeric N-terminal hydrophobic fusion peptide of the ectodomain of HIV-1 gp41 with a trimeric (pII) variant of the GCN4 coiled coil region produced a crystallizable protein that retained structural features of native gp41 [41]. There are over 33 000 single-span membrane proteins in the UniProt database (http://www.uniprot.org/locations/SL-9904), the full-length structures of the vast majority of which are not yet known. Our ‘solubilization by design’ approach using the GCN4 leucine zipper motif in place of a single TM helix may prove useful for overproduction of soluble forms of many dimeric, trimeric and tetrameric single-span membrane proteins for structural and functional studies.
Acknowledgements
Genomic DNA of strain 26695 H. pylori was a kind gift of Dr Terry Kwok-Schuelein (Monash University). We acknowledge and thank Dr Marcel Hijnen (Monash University) for assistance with MALLS/QELS analysis and A/Prof. Martin Stone (Monash University) for advice on chimaera design and helpful discussion. This work was supported by the Australian Research Council (ARC DP1094619, to A.R.). A.R. is an ARC Research Fellow and M.C.W. is an NHMRC Senior Research Fellow.
References
- 1.Ottemann KM, Miller JF. 1997. Roles for motility in bacterial–host interactions. Mol. Microbiol. 24, 1109–1117 10.1046/j.1365-2958.1997.4281787.x (doi:10.1046/j.1365-2958.1997.4281787.x) [DOI] [PubMed] [Google Scholar]
- 2.Ottemann KM, Lowenthal AC. 2002. Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect. Immun. 70, 1984–1990 10.1128/IAI.70.4.1984-1990.2002 (doi:10.1128/IAI.70.4.1984-1990.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blair DF, Kim DY, Berg HC. 1991. Mutant MotB proteins in Escherichia coli. J. Bacteriol. 173, 4049–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josenhans C, Suerbaum S. 2002. The role of motility as a virulence factor in bacteria. Int. J. Med. Microbiol. 291, 605–614 10.1078/1438-4221-00173 (doi:10.1078/1438-4221-00173) [DOI] [PubMed] [Google Scholar]
- 5.Minamino T, Imada K, Namba K. 2008. Molecular motors of the bacterial flagella. Curr. Opin. Struct. Biol. 18, 693–701 10.1016/j.sbi.2008.09.006 (doi:10.1016/j.sbi.2008.09.006) [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Lin T, Botkin DJ, McCrum E, Winkler H, Norris SJ. 2009. Intact flagellar motor of Borrelia burgdorferi revealed by cryo-electron tomography: evidence for stator ring curvature and rotor/C-ring assembly flexion. J. Bacteriol. 191, 5026–5036 10.1128/JB.00340-09 (doi:10.1128/JB.00340-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macnab RM. 2004. Type III flagellar protein export and flagellar assembly. Biochim. Biophys. Acta 1694, 207–217 10.1016/j.bbamcr.2004.04.005 (doi:10.1016/j.bbamcr.2004.04.005) [DOI] [PubMed] [Google Scholar]
- 8.Reid SW, Leake MC, Chandler JH, Lo CJ, Armitage JP, Berry RM. 2006. The maximum number of torque-generating units in the flagellar motor of Escherichia coli is at least 11. Proc. Natl Acad. Sci. USA 103, 8066–8071 10.1073/pnas.0509932103 (doi:10.1073/pnas.0509932103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kojima S, Blair DF. 2004. Solubilization and purification of the MotA/MotB complex of Escherichia coli. Biochemistry 43, 26–34 10.1021/bi035405l (doi:10.1021/bi035405l) [DOI] [PubMed] [Google Scholar]
- 10.Berg HC. 2003. The rotary motor of bacterial flagella. Annu. Rev. Biochem. 72, 19–54 10.1146/annurev.biochem.72.121801.161737 (doi:10.1146/annurev.biochem.72.121801.161737) [DOI] [PubMed] [Google Scholar]
- 11.Braun TF, Al-Mawsawi LQ, Kojima S, Blair DF. 2004. Arrangement of core membrane segments in the MotA/MotB proton-channel complex of Escherichia coli. Biochemistry 43, 35–45 10.1021/bi035406d (doi:10.1021/bi035406d) [DOI] [PubMed] [Google Scholar]
- 12.Manson M, Tedesco P, Berg H, Harold F, van der Drift C. 1977. A protonmotive force drives bacterial flagella. Proc. Natl Acad. Sci. USA 74, 3060–3064 10.1073/pnas.74.7.3060 (doi:10.1073/pnas.74.7.3060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roujeinikova A. 2008. Crystal structure of the cell wall anchor domain of MotB, a stator component of the bacterial flagellar motor: implications for peptidoglycan recognition. Proc. Natl Acad. Sci. USA 105, 10 348–10 353 10.1073/pnas.0803039105 (doi:10.1073/pnas.0803039105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reboul CF, Andrews DA, Nahar MF, Buckle AM, Roujeinikova A. 2011. Crystallographic and molecular dynamics analysis of loop motions unmasking the peptidoglycan-binding site in stator protein MotB of flagellar motor. PLoS ONE 6, e18981. 10.1371/journal.pone.0018981 (doi:10.1371/journal.pone.0018981) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Neill J, Xie M, Hijnen M, Roujeinikova A. 2011. Role of the MotB linker in the assembly and activation of the bacterial flagellar motor. Acta Crystallogr. D 67, 1009–1016 10.1107/S0907444911041102 (doi:10.1107/S0907444911041102) [DOI] [PubMed] [Google Scholar]
- 16.Kojima S, Imada K, Sakuma M, Sudo Y, Kojima C, Minamino T, Homma M, Namba K. 2009. Stator assembly and activation mechanism of the flagellar motor by the periplasmic region of MotB. Mol. Microbiol. 73, 710–718 10.1111/j.1365-2958.2009.06802.x (doi:10.1111/j.1365-2958.2009.06802.x) [DOI] [PubMed] [Google Scholar]
- 17.Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, Stemmler TL, Mobashery S. 2006. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl Acad. Sci. USA 103, 4404–4409 10.1073/pnas.0510182103 (doi:10.1073/pnas.0510182103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Shea EK, Klemm JD, Kim PS, Alber T. 1991. X-Ray structure of the GCN4 leucine zipper, a 2-stranded, parallel coiled coil. Science 254, 539–544 10.1126/science.1948029 (doi:10.1126/science.1948029) [DOI] [PubMed] [Google Scholar]
- 19.Hosking ER, Vogt C, Bakker EP, Manson MD. 2006. The Escherichia coli MotAB proton channel unplugged. J. Mol. Biol. 364, 921–937 10.1016/j.jmb.2006.09.035 (doi:10.1016/j.jmb.2006.09.035) [DOI] [PubMed] [Google Scholar]
- 20.Braun TF, Blair DF. 2001. Targeted disulfide cross-linking of the MotB protein of Escherichia coli: evidence for two H+ channels in the stator complex. Biochemistry 40, 13 051–13 059 10.1021/bi011264g (doi:10.1021/bi011264g) [DOI] [PubMed] [Google Scholar]
- 21.Wilson ML, Macnab RM. 1990. Co-overproduction and localization of the Escherichia coli motility proteins MotA and MotB. J. Bacteriol. 172, 3932–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Neill J, Roujeinikova A. 2008. Cloning, purification and crystallization of MotB, a stator component of the proton-driven bacterial flagellar motor. Acta Crystallogr. F 64, 561–563 10.1107/S1744309108012219 (doi:10.1107/S1744309108012219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomb JF, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547 10.1038/41483 (doi:10.1038/41483) [DOI] [PubMed] [Google Scholar]
- 24.Andrade MA, Chacón P, Merelo JJ, Morán F. 1993. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6, 383–390 10.1093/protein/6.4.383 (doi:10.1093/protein/6.4.383) [DOI] [PubMed] [Google Scholar]
- 25.Orwig SD, Lieberman RL. 2011. Biophysical characterization of the olfactomedin domain of myocilin, an extracellular matrix protein implicated in inherited forms of glaucoma. PLoS ONE 6, e16347. 10.1371/journal.pone.0016347 (doi:10.1371/journal.pone.0016347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. 2006. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst. 39, 277–286 10.1107/S0021889806004699 (doi:10.1107/S0021889806004699) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 36, 1277–1282 10.1107/S0021889803012779 (doi:10.1107/S0021889803012779) [DOI] [Google Scholar]
- 28.Svergun D. 1992. Determination of the regularization parameter in indirect transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 10.1107/S0021889892001663 (doi:10.1107/S0021889892001663) [DOI] [Google Scholar]
- 29.Mylonas E, Svergun DI. 2007. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Cryst. 40, s245–s249 10.1107/S002188980700252X (doi:10.1107/S002188980700252X) [DOI] [Google Scholar]
- 30.Ortega A, Amoros D, de la Torre JD. 2011. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 10.1016/j.bpj.2011.06.046 (doi:10.1016/j.bpj.2011.06.046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geourjon C, Deléage G. 1995. SOPMA: significant improvement in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684 10.1093/bioinformatics/11.6.681 (doi:10.1093/bioinformatics/11.6.681) [DOI] [PubMed] [Google Scholar]
- 32.Feigin LA, Svergun DI. 1987. Analysis by small-angle X-ray and neutron scattering, pp. 83–87 New York, NY: Plenum Press [Google Scholar]
- 33.Cochran AG, Kim PS. 1996. Imitation of Escherichia coli aspartate receptor signaling in engineered dimers of the cytoplasmic domain. Science 271, 1113–1116 10.1126/science.271.5252.1113 (doi:10.1126/science.271.5252.1113) [DOI] [PubMed] [Google Scholar]
- 34.Frank S, Kammerer RA, Hellstern S, Pegoraro S, Stetefeld J, Lustig A, Moroder L, Engel J. 2000. Toward a high-resolution structure of phospholamban: design of soluble transmembrane domain mutants. Biochemistry 39, 6825–6831 10.1021/bi0000972 (doi:10.1021/bi0000972) [DOI] [PubMed] [Google Scholar]
- 35.Li H, Cocco MJ, Steitz TA, Engelman DE. 2001. Conversion of phospholamban into a soluble pentameric helical bundle. Biochemistry 40, 6636–6645 10.1021/bi0026573 (doi:10.1021/bi0026573) [DOI] [PubMed] [Google Scholar]
- 36.Slovic AM, Kono H, Lear JD, Saven JG, DeGrado WF. 2004. Computational design of water-soluble analogues of the potassium channel KcsA. Proc. Natl Acad. Sci. USA 101, 1828–1833 10.1073/pnas.0306417101 (doi:10.1073/pnas.0306417101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stefer S, et al. 2011. Structural basis for tail-anchored membrane protein biogenesis by the Get3-receptor complex. Science 333, 758–762 10.1126/science.1207125 (doi:10.1126/science.1207125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matias VR, Al-Amoudi A, Dubochet J, Beveridge TJ. 2003. Cryo-transmission electron microscopy of frozen-hydrated sections of Escherichia coli and Pseudomonas aeruginosa. J. Bacteriol. 185, 6112–6118 10.1128/JB.185.20.6112-6118.2003 (doi:10.1128/JB.185.20.6112-6118.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woolfson DN. 2005. The design of coiled-coil structures and assemblies. Adv. Protein Chem. 70, 79–112 10.1016/S0065-3233(05)70004-8 (doi:10.1016/S0065-3233(05)70004-8) [DOI] [PubMed] [Google Scholar]
- 40.Djuranovic S, Hartmann MD, Habeck M, Ursinus A, Zwickl P, Martin J, Lupas AN, Zeth K. 2009. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol. Cell. 34, 580–590 10.1016/j.molcel.2009.04.030 (doi:10.1016/j.molcel.2009.04.030) [DOI] [PubMed] [Google Scholar]
- 41.Weissenhorn W, Calder LJ, Dessen A, Laue T, Skehel JJ, Wiley DC. 1997. Assembly of a rod-shaped chimaera of a trimeric GCN4 zipper and the HIV-1 gp41 ectodomain expressed in Escherichia coli. Proc. Natl Acad. Sci. USA 94, 6065–6069 10.1073/pnas.94.12.6065 (doi:10.1073/pnas.94.12.6065) [DOI] [PMC free article] [PubMed] [Google Scholar]