

Abstract
The theory of the clonal origin of cancer states that a tumour arises from one cell that acquires mutation(s) leading to the malignant phenotype. It is the current belief that many of these mutations give a fitness advantage to the mutant population allowing it to expand, eventually leading to disease. However, mutations that lead to such a clonal expansion need not give a fitness advantage and may in fact be neutral—or almost neutral—with respect to fitness. Such mutant clones can be eliminated or expand stochastically, leading to a malignant phenotype (disease). Mutations in haematopoietic stem cells give rise to diseases such as chronic myeloid leukaemia (CML) and paroxysmal nocturnal haemoglobinuria (PNH). Although neutral drift often leads to clonal extinction, disease is still possible, and in this case, it has important implications both for the incidence of disease and for therapy, as it may be more difficult to eliminate neutral mutations with therapy. We illustrate the consequences of such dynamics, using CML and PNH as examples. These considerations have implications for many other tumours as well.
Keywords: cancer, stem cells, mathematical models
1. Introduction
Tissue homeostasis gives the impression that cell populations are constant and hides the fact that beneath this appearance of stability, there is substantial cell turnover. This is the case in all epithelial tissues (skin, gut, breast) as well as in haematopoiesis. These tissues have a similar architecture where at the root lie tissue-specific stem cells that divide to self-renew and give rise to progeny cells that differentiate into the various types of cells present within the tissue. As long as the dynamic exchanges between these populations are balanced, homeostasis ensues. Therefore, at some macroscopic timescale, the sizes of these hierarchically organized populations, arising from the stem cell population, appear constant.
The stochastic dynamics in such stem cell populations of constant size can conveniently be modelled by the Moran process from population genetics [1–3], a birth–death process (figure 1). Each cell has a certain fitness that is in our case constant. In each time step, a cell is chosen for reproduction at random, but with probability proportional to the fitness. This cell produces identical offspring, replacing a randomly chosen cell, which is considered to differentiate to the next stage. Effectively, this is a death event, because that cell leaves the stem cell compartment and cannot be selected again for reproduction. In this model, cell export captures the initial step in the path towards differentiation of that cell, usually through intermediate cells known as progenitors. Stem cells can acquire mutations, and it is possible that these confer a higher reproductive fitness to the mutated stem cells, hence increasing the chances that the mutant population will invade the whole stem cell compartment. This leads to a so-called clone that corresponds to all the offspring of the mutant cell in all further differentiation steps. Other mutations may give a fitness disadvantage and such a clone will be eliminated with a probability that increases with the population size and the fitness disadvantage of the cell. As such, these two extremes will induce two potential outcomes: either the mutant population takes over completely or it undergoes extinction. Additionally, certain mutations may not cause a reproductive advantage, i.e. neutral mutations. This case is also a good approximation if selection is weak, i.e. if the product of population size and fitness differences is small [4]. Even in this case, the potential outcomes will, in the long run, be the same as for the two extreme cases mentioned earlier.
Figure 1.

Possible changes of the number of mutated cells in an elementary time step of the Moran process. (a) The number of mutated cells can increases by one if a wild-type cell differentiates and a mutated cell divides. (b,c) If the same cell type is affected by cell division and differentiation, then the number of mutated cells does not change. (d) The number of mutated cells decreases by one if a mutated cell differentiates and is replaced by a wild-type cell. (Online version in colour.)
However, all organisms have a finite lifetime and so one expects intermediate regimes with mixed populations. Such mixed regimes last longest for neutral mutations when fitness is constant [5]. Such a scenario is not a mere theoretical exercise. Many mutations are indeed neutral [6,7] but the impact of a mutation is cell-context-dependent. In fact, the same mutation may be neutral in one microenvironment but exhibit a fitness difference in another [8–10]. Fitness may also fluctuate in time [11], reflecting changes in the environment of the cells. Here, we do not consider this more complex scenario, but concentrate on fixed fitness values.
There is some evidence, for example, that BCR-ABL expression in a haematopoietic stem cell (HSC) gives no fitness advantage to the cell while the same oncogene expressed in progenitor cells confers to them a fitness advantage [9,10,12–14]. Therefore, the expansion of a BCR-ABL mutant stem cell clone could rely entirely on neutral drift. In a similar manner, neutral drift can explain the incidence, population age structure and spontaneous elimination of PIG-A mutant clones [15,16], a gene required for the synthesis of glycosylphosphatidyl inositol-linked proteins that leads to the phenotype observed in paroxysmal nocturnal haemoglobinuria (PNH) [17–19]. In fact, the change of the reproductive fitness of cells owing to acquired mutations is often unrelated to their effect on the entire organism where ‘disease’ may or may not manifest itself as a reduction in the ‘fitness’ of the entire organism.
Let us consider the appearance of a mutation, say BCR-ABL, that has no impact on the reproduction of the HSCs [12–14,20]. Given that the size of the active HSC pool (i.e. the number of HSC that are contributing to haematopoiesis) is small (of the order of 400 cells [21,22]), one expects stochastic effects to have a prominent impact on the clonal evolution of the mutant population [2,23], as also observed in related systems [24]. The theory of the clonal origin of cancer tells us that we start with one cell that arises owing to mutation [25]. How will this clone evolve in time? If the first mutant cell is selected for export, the mutation will disappear from the HSC pool, and only a new mutation will re-initiate such a lineage, an unlikely event [26,27]. By contrast, if such a cell is selected for reproduction, it will generate another BCR-ABL expressing cell (because back mutation to a normal state is a very unlikely scenario). As the population of mutant cells increases, the probability that one of them is selected either for reproduction or export also increases. What would be the rate of expansion of such a population? This process is relevant, because persistence of a ‘neutral’ clone can maintain disease in the absence of therapy that can specifically kill such cells [10,19,28]. Moreover, such a process may in part explain why in many tumours, the cancer stem cell population is small, while in others, many of the cancer cells have tumour initiating capability [29,30]. In the following, we provide a mathematical description of such a process and show how stochastic simulations (see appendix) confirm such a behaviour.
2. Stem cell dynamics
Consider a population of constant size N, where cells replicate and differentiate in discrete time steps. In each time step, a cell is selected for reproduction, proportional to fitness and divides symmetrically, producing identical offspring. In the neutral case with j mutant cells, the probability that we select one of them for reproduction is j/N, while with probability (N−j)/N, we select a normal cell for reproduction; see table 1 for an overview of the notation. If the mutant cells have a fitness r, the probability of selection is rj/(rj + N−j). If r > 1, the mutant has a higher relative fitness compared with the wild-type. If r < 1, the mutant has a lower relative fitness, whereas if r = 1, mutant and normal cells have the same fitness. Given that the total population of cells remains constant, then with each reproduction event, we choose a cell for export at random—a typical assumption is that this cell differentiates during cell division and cannot be considered as a primitive stem cell anymore. With probability j/N, we choose a mutant cell for export, whereas with probability (N−j)/N, we choose a normal cell for export. When the process is repeated N times, on average, each cell would have had one chance to reproduce. This natural timescale of the process is often referred to as generation. For example, in the case of the active HSC pool, we have N = 400, and each HSC reproduces, on average, about once per year [21,22]. Therefore, when 400 selection–reproduction–export events have occurred, a year will have passed and, on average, each cell would have reproduced once (for neutral mutations).
Table 1.
Overview of symbols used.
| symbol | description |
|---|---|
| N | population size (fixed) |
| r | relative fitness of the mutated cell type |
| t | time step, N time steps are one generation |
| j,k | number of mutants |
| Tj→k | probability to go from j to k in one time step |
| Mj→kt | probability to go from j to k in t time steps |
| ct | number of mutants at time t conditioned upon no extinction, starting from 1 |
| 〈ct〉 | expected number of mutants at time t conditioned upon no extinction, starting from 1 |
| 〈ct2〉 | expected of the square of ct |
Note that we have excluded the possibility that a cell divides asymmetrically and produces one differentiated daughter cell and one daughter cell identical to the parent cell. The presence of such asymmetric stem cell divisions would slow down the dynamics between different stem cells. If all cell divisions would be asymmetric, the number of stem cells of each type would remain constant, with no room for expansion or extinction of mutated stem cells within the stem cell pool.
2.1. Moran process
The neutral Moran process is a birth–death process with transition probabilities from state j to state k
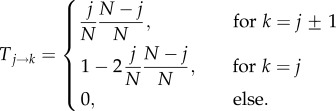 |
2.1 |
(figure 1). Let us first address the probability that the mutant clone goes extinct. For a neutral mutation, this probability is given by 1−N−1 in the long run. For short times, we can add the different paths that lead to extinction. The number of such possible paths increases rapidly with t: there is only one path to extinction in one time step or two time steps, there are two paths to extinction in three time steps, and there are four paths to extinction in four time steps. For example, the probability that the mutant cells go extinct within t = 4 time steps is given by
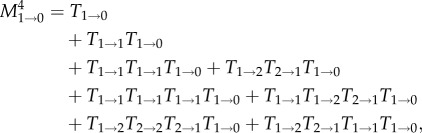 |
2.2 |
where we have introduced the transition matrix after t time steps, Mi→jt = (Ti→j)t for notational convenience. Let us now approximate M1→0t for large N. Because j≪N, we have 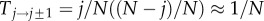 and
and 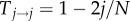
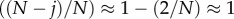 for j≪N. The probability to stay in a state is approximately 1, but the probability to move to an adjacent state is close to zero. Thus, we can approximate M1→0t by considering the paths that have up to only a certain number of transitions between states. Taking into account only transitions in which the number of mutants changes only once from one to zero, we obtain
for j≪N. The probability to stay in a state is approximately 1, but the probability to move to an adjacent state is close to zero. Thus, we can approximate M1→0t by considering the paths that have up to only a certain number of transitions between states. Taking into account only transitions in which the number of mutants changes only once from one to zero, we obtain
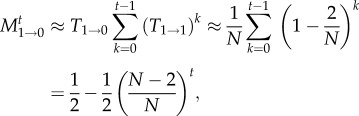 |
2.3 |
where the approximation is valid for large N. For t≫N, the result can be approximated by t/N. We can improve this approximation by taking into account also those paths in which we reach extinction with up to three transitions between states, which yields for t ≥ 3 the improved approximation
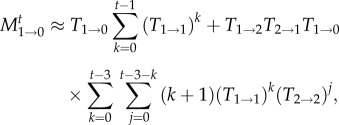 |
2.4 |
Going one step further, we could also include the paths involving five transitions, which would lead to two additional terms for t ≥ 5,
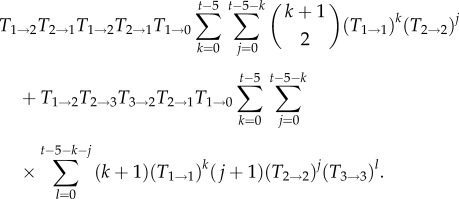 |
Note that there are two classes of paths, one in which the state with two mutants is entered and left twice (first line) and one in which the state with three mutants is reached (second line). However, figure 2 illustrates that this approximation is only a marginal improvement over the three-transition approximation equation (2.4), which indicates that to derive a better approximation of the time-dependent extinction probability for larger times, we would need to consider a very large number of terms. A similar effect is found for the Wright–Fisher process [31,32].
Figure 2.

Time-dependent probability of extinction. We show the analytical approximations based on equations (2.3)–(2.5) and simulations for N = 100 (circles with error bars given by the standard error of the binomial distribution) and for N = 10 000 (line). Time has been re-scaled to generations, one generation consists of N time steps. For the simulations, the population size has a marginal influence only for N > 100. While the approximation with three transitions, equation (2.4) is a significant improvement over the approximation with only one transition, equation (2.3), taking into account five transitions (equation (2.5)) does not improve the approximation much further. This indicates that for a better approximation for long times, many terms have to be taken into account (simulations averaged over 100 realizations). (Online version in colour.)
We are also interested in the conditional average number of mutants c after t time steps given that the mutants do not go extinct, 〈ct〉. This can be obtained by averaging only over those paths that do not lead to state k = 0,
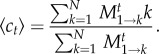 |
2.5 |
For the numerator, we have at t = 1
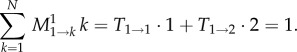 |
2.6 |
Thus, the average number of mutants does not change in the first time step. Because the transition probabilities are constant in time, it does not change in the second time step either. This can be iterated to see that the average number of mutants remains constant (this can also be seen directly from the transition probabilities: for neutral processes, Ti→i−1 = Ti→i+1 and thus, the average cannot change—see also the discussion of the branching process in the appendix). Thus, 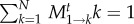 . In the denominator of equation (2.5), the transition probabilities out of state 1 are normalized. In other words, after t time steps, the process has to end up somewhere between 0 and N. Thus, the probabilities to end up in a state k > 0 or in state k = 0 must sum up to one,
. In the denominator of equation (2.5), the transition probabilities out of state 1 are normalized. In other words, after t time steps, the process has to end up somewhere between 0 and N. Thus, the probabilities to end up in a state k > 0 or in state k = 0 must sum up to one, 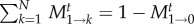 . The term M1→0t has already been calculated above. With this, we have for t≪N
. The term M1→0t has already been calculated above. With this, we have for t≪N
| 2.7 |
Figure 3 shows that this linear increase is a good approximation not only for small t, but for the first few generations. This is consistent with the arguments above. Because the conditional fixation time of a single neutral mutant is N−1 generations [33–35], we expect that after 2N generations, the conditional number of mutants is already close to N, as seen in our simulations for N = 10 in figure 3 (see appendix for an explanation of the simulations).
Figure 3.

The average fraction of mutants conditioned upon no extinction. Our analytical approximation equation (2.7) is good for up to N/4 generations, in contrast to the approximations for the probability of extinction, which holds only for a single generation (cf. figure 2). For longer times, the fraction of mutants increases at a slower rate, reaching the maximum N after approximately 2N generations, which is consistent with the average conditional fixation time of the neutral mutants. The distribution becomes broader with time, but narrows again when almost all realizations have reached fixation, as illustrated for the case of N = 10 (error bars represent the standard deviation obtained from averages over 106 independent realizations). (Online version in colour.)
In order to infer the variance of ct, we calculate the quantity
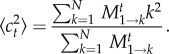 |
2.8 |
Again, we apply the same approximation as above and consider only those terms that are based on at most a single transition between states; we find for the numerator of equation (2.8)
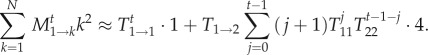 |
2.9 |
Note that this approximation neglects transitions to states k > 2. The next term, in which two transitions are allowed, would lead to two additional terms,
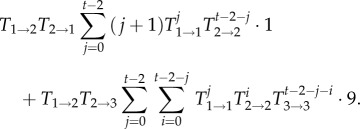 |
2.10 |
However, including this term leads only to a minor improvement of the approximation. Next, we approximate equation (2.9) for large N in the form 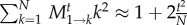 , and find
, and find
| 2.11 |
Finally, the error of our quantity 〈ct〉 can be approximated by
| 2.12 |
Thus, the leading dependence in t of our approximation is linear, in accord with the numerical results shown in figure 3. Note, however, that our approximation for the error holds quantitatively for very short times only. We note that the limit of the approximation is not the Taylor expansion for large N, but the assumption that only few transitions occur. For more precise approximations, a large number of such terms would have to be taken into account.
3. Discussion
The results of our simulations and mathematical analysis have implications for the incidence of acquired HSC disorders. Given that a population with a neutral mutation (or a mutation that confers a very small change in fitness, N(r−1)≪1), on average increases by only one cell per year, then we can conclude that in a disease such as PNH, the original mutation in PIG-A must occur early in life for the clone to reach a threshold compatible with disease (e.g. 10% of the stem cell population being mutated [36]), given that the average age of patients with this disease is in the fourth or fifth decade of life. Of course, this is the average and in principle, growth of the population can be significantly faster—or slower—owing to stochastic effects alone [2]. The other option would be to imply some sort of fitness advantage for the clone either owing to an intrinsic effect, as for instance a second mutation, such as HMGA2 [18,37], or extrinsic effect such as an immune-mediated attack on normal HSC as proposed by Luzzatto et al. [38]. As discussed elsewhere [26], the probability of a second mutation in the same cell that gives it a fitness advantage is small, and perhaps this is the reason why to date, only a couple of patients have been described with such a second mutation [18]. On the other hand, a fitness advantage could explain instances where the clone is very large, while neutral drift would explain the majority of situations where the clone is large enough to cause disease [19].
In the case of CML, the incidence is almost invariably in people beyond the second or third decade of life. The latent period between the appearance of the Philadelphia chromosome [39] in a HSC and diagnosis is often considered to be in the range of 3–5 years. Therefore, it appears that most mutations leading to the Ph' chromosome occur later in life, presumably because the probability of a chromosomal recombination event leading to such a specific translocation is smaller than the probability of a mutation in PIG-A where a variety of mutations can inactivate the gene [40–42].
Our modelling can also explain an apparent paradox: many mutations in PIG-A inactivate the gene but clinically significant PNH is rare. CML is a significantly more common disorder compared with PNH (by at least one order of magnitude), despite the fact that it is triggered by only a single mutation. Both diseases start with a single mutated cell. Neutral drift means that clonal expansion, in general will be slow and this is enough to explain why PNH is rare. If BCR-ABL expression does not give a fitness advantage, why is the incidence of the disease significantly more common? The difference lies in the fact that BCR-ABL expression does give a fitness advantage to cells downstream of the HSC owing to enhanced self-renewal of the cells [10,27,28,43], leading to their expansion with myeloproliferation and ultimately disease. Hence, and although the population of CML stem cells is small, the disease is driven by progenitor cells that rapidly expand in number and essentially may invade haematopoiesis leading to disease. If CML stem cells would instead have an increased propensity to differentiate, the clone would die out faster, because increased differentiation implies loss of these cells from the stem cell compartment.
An alternative possibility would be that chromosomal instability induced by BCR-ABL expression [44,45] would result in the accumulation of additional mutations that can accelerate the development of disease. This line of reasoning implies that BCR-ABL expression alone may not be enough to explain the initial development of chronic-phase CML. In our view, this hypothesis would not be in keeping with the results of animal experiments where a disease phenotype similar to chronic phase CML is induced within weeks of the introduction of BCR-ABL expressing HSC in immunodeficient animals [46]. However, even in this case, the affected cells would need a fitness advantage to lead to an early onset of the disease compared with the neutral case. Note that our model does not capture late stages of the disease, where additional mutations may lead to different phenotypes. In addition, in these stages, the assumption of a constant population size may break down.
CML can be well controlled, and haematopoiesis returns to normal because at the level of the stem cell pool, the majority of cells are normal [47] and haematopoiesis recovers once the impact of BCR-ABL on cells is blocked by tyrosine kinase inhibitors such as imatinib. By contrast, expansion of the clone in PNH is less likely, because PIG-A does not give a fitness advantage either to HSC or progenitor cells. Moreover, as shown earlier, when PNH is diagnosed, reflecting the (less likely) success of clonal expansion, the clone size at the level of the stem cell pool will in general be sizeable, which makes elimination of the clone extremely difficult—except for stochastic extinction [17,19], only a bone marrow transplant will eliminate the clone and cure the disease. Therapy with eculizumab, a monoclonal antibody that inhibits the C5 complement component and prevents intravascular haemolysis, has no impact on clone size in this disease, and therefore is not curative [48,49].
As can be seen from this analysis, neutral mutations have many flavours with implications for a variety of disorders that ostensibly arise within the same cell. As in real estate, location is everything. A neutral mutation in some cells may provide a fitness advantage to their progeny cells (BCR-ABL) that can also make them amenable to therapy. A neutral mutation may be an innocuous passenger but at times leads to a disease that can be as resistant as any other with respect to therapy. Small may not be cute—it may be stubborn, even lethal.
These evolutionary considerations have potential implications for other clonal disorders as well. Genomic sequencing of tumours identifies many mutated genes. The majority of these mutated genes are thought to be ‘passengers’ [50], perhaps implying that they are not important for tumour growth or survival. Such nomenclature may be misleading, as potentially important mutations that are relevant or even critical for the tumour may be ignored.
Appendix
A.1. Branching process
To connect our work to previous papers based on branching processes, we can also consider the dynamics of mutant cells as a branching process [51–53]. As a timescale, we consider a single cell division. With probability p = 1/N, a particular cell produces two offspring cells. With the same probability p, it is exported and lost from the stem cell pool (this is equivalent to asymmetric cell division). Finally, with probability 1−2p, it remains unaffected (and thus can be considered as having a single offspring), the equivalent of self-renewal. Thus, the average number of offspring at time t is given by 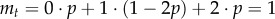 , which implies a constant average number of mutated stem cells. Owing to this restriction, we are dealing with a critical branching process [52]. This implies that the mutated stem cells eventually go extinct with probability one. However, because we consider a population of size N, once the mutated cells hit this threshold (i.e. they invade the population) they cannot go extinct anymore. But such a statement is not necessarily helpful in our context, because extinction may take longer than the lifetime of the associated individual. Because extinction is likely, the fact that the average number of mutated stem cells is constant implies large fluctuations of the process [2,52]. The variance in the number of offspring is given by
, which implies a constant average number of mutated stem cells. Owing to this restriction, we are dealing with a critical branching process [52]. This implies that the mutated stem cells eventually go extinct with probability one. However, because we consider a population of size N, once the mutated cells hit this threshold (i.e. they invade the population) they cannot go extinct anymore. But such a statement is not necessarily helpful in our context, because extinction may take longer than the lifetime of the associated individual. Because extinction is likely, the fact that the average number of mutated stem cells is constant implies large fluctuations of the process [2,52]. The variance in the number of offspring is given by 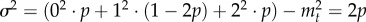 . From the variance, we can obtain the scaling of the expected number of mutated cells ct given that
. From the variance, we can obtain the scaling of the expected number of mutated cells ct given that 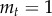 ,
, 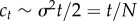 [52]. This scaling holds for large t. Thus, the expected number of mutated stem cells given that they do not go extinct grow by one per generation. This has also been observed in a Moran model of stem cell dynamics [28]. Next, we show that the result can also be obtained directly from the Moran model when the population size is not too small. This illustrates that the conditional growth rate of 1 per generation is valid even for short time t.
[52]. This scaling holds for large t. Thus, the expected number of mutated stem cells given that they do not go extinct grow by one per generation. This has also been observed in a Moran model of stem cell dynamics [28]. Next, we show that the result can also be obtained directly from the Moran model when the population size is not too small. This illustrates that the conditional growth rate of 1 per generation is valid even for short time t.
A.2. Computer simulations
For the computer simulations, the Moran process was implemented as follows: the system is characterized by the number of mutants j. In each time step, the probabilities to transit to j±1 or to stay in j are calculated based on j, N and r, see §2. Then, a single pseudorandom number is generated that determines which of the three possible transitions is realized (transfer to j±1 or stay in j). The procedure is then continued in the next state. For the time-dependent extinction probability, the fraction of runs leading to the extinction of all mutants until a certain time is computed (figure 2). For the number of cells given no extinction, as shown in figure 3, only those runs in which extinction did not occur so far are considered.
References
- 1.Moran PAP. 1962. The statistical processes of evolutionary theory. Oxford, UK: Clarendon Press [Google Scholar]
- 2.Dingli D, Traulsen A, Pacheco JM. 2007. Stochastic dynamics of haematopoietic tumor stem cells. Cell Cycle 6, 461–466 10.4161/cc.6.4.3853 (doi:10.4161/cc.6.4.3853) [DOI] [PubMed] [Google Scholar]
- 3.Werner B, Lutz D, Brümmendorf TH, Traulsen A, Balabanov S. 2011. Dynamics of resistance development to imatinib under increasing selection pressure: a combination of mathematical models and in vitro data. PLoS ONE 6, e28955. 10.1371/journal.pone.0028955 (doi:10.1371/journal.pone.0028955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komarova NL, Sengupta A, Nowak MA. 2003. Mutation–selection networks of cancer initiation: tumor suppressor genes and chromosomal instability. J. Theor. Biol. 223, 433–450 10.1016/S0022-5193(03)00120-6 (doi:10.1016/S0022-5193(03)00120-6) [DOI] [PubMed] [Google Scholar]
- 5.Antal T, Scheuring I. 2006. Fixation of strategies for an evolutionary game in finite populations. Bull. Math. Biol. 68, 1923–1944 10.1007/s11538-006-9061-4 (doi:10.1007/s11538-006-9061-4) [DOI] [PubMed] [Google Scholar]
- 6.Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217, 624–626 10.1038/217624a0 (doi:10.1038/217624a0) [DOI] [PubMed] [Google Scholar]
- 7.Kimura M. 1986. DNA and the neutral theory. Phil. Trans. R. Soc. Lond. B 312, 343–354 10.1098/rstb.1986.0012 (doi:10.1098/rstb.1986.0012) [DOI] [PubMed] [Google Scholar]
- 8.Dingli D, Antal T, Traulsen A, Pacheco JM. 2009. Progenitor cell self-renewal and cyclic neutropenia. Cell Prolif. 42, 330–338 10.1111/j.1365-2184.2009.00598.x (doi:10.1111/j.1365-2184.2009.00598.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Traulsen A, Pacheco JM, Dingli D. 2010. Reproductive fitness advantage of BCR-ABL expressing leukemia cells. Cancer Lett. 294, 43–48 10.1016/j.canlet.2010.01.020 (doi:10.1016/j.canlet.2010.01.020) [DOI] [PubMed] [Google Scholar]
- 10.Dingli D, Traulsen A, Lenaerts T, Pacheco JM. 2010. Evolutionary dynamics of chronic myeloid leukemia. Genes Cancer 1, 309–315 10.1177/1947601910371122 (doi:10.1177/1947601910371122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell G. 2010. Fluctuating selection: the perpetual renewal of adaptation in variable environments. Phil. Trans. R. Soc. B 365, 87–97 10.1098/rstb.2009.0150 (doi:10.1098/rstb.2009.0150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marley SB, Gordon MY. 2005. Chronic myeloid leukaemia: stem cell derived but progenitor cell driven. Clin. Sci. 109, 13–25 10.1042/CS20040336 (doi:10.1042/CS20040336) [DOI] [PubMed] [Google Scholar]
- 13.Schemionek M. et al. 2010. BCR-ABL enhances differentiation of long-term repopulating hematopoietic stem cells. Blood 115, 3185–3195 10.1182/blood-2009-04-215376 (doi:10.1182/blood-2009-04-215376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Järås M, et al. 2010. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl Acad. Sci. USA 107, 16280–16285. 10.1073/pnas.1004408107 (doi:10.1073/pnas.1004408107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luzzatto L, Bessler M, Rotoli B. 1997. Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise? Cell 88, 1–4 10.1016/S0092-8674(00)81850-4 (doi:10.1016/S0092-8674(00)81850-4) [DOI] [PubMed] [Google Scholar]
- 16.Araten DJ, Luzzatto L. 2006. The mutation rate in PIG-A is normal in patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood 108, 734–736 10.1182/blood-2006-01-0256 (doi:10.1182/blood-2006-01-0256) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. 1995. Natural history of paroxysmal nocturnal hemoglobinuria. New Engl. J. Med. 333, 1253–1259 10.1056/NEJM199511093331904 (doi:10.1056/NEJM199511093331904) [DOI] [PubMed] [Google Scholar]
- 18.Inoue N. et al. 2006. Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood 108, 4232–4236 10.1182/blood-2006-05-025148 (doi:10.1182/blood-2006-05-025148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dingli D, Luzzatto L, Pacheco JM. 2008. Neutral evolution in paroxysmal nocturnal hemoglobinuria. Proc. Natl Acad. Sci. USA 105, 18 496–18 500. 10.1073/pnas.0802749105 (doi:10.1073/pnas.0802749105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. 2011. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 121, 396–409 10.1172/JCI35721 (doi:10.1172/JCI35721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buescher ES, Alling DW, Gallin JI. 1985. Use of an X-linked human neutrophil marker to estimate timing of lyonization and size of the dividing stem cell pool. J. Clin. Invest. 76, 1581–1584 10.1172/JCI112140 (doi:10.1172/JCI112140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dingli D, Pacheco JM. 2006. Allometric scaling of the active hematopoietic stem cell pool across mammals. PLoS ONE 1, e2. 10.1371/journal.pone.0000002 (doi:10.1371/journal.pone.0000002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abkowitz JL, Catlin SN, Guttorp P. 1996. Evidence that hematopoiesis may be a stochastic process in vivo. Nat. Med. 2, 190–197 10.1038/nm0296-190 (doi:10.1038/nm0296-190) [DOI] [PubMed] [Google Scholar]
- 24.Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. 2012. Defining the mode of tumour growth by clonal analysis. Nature 488, 527–530 10.1038/nature11344 (doi:10.1038/nature11344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cairns J. 1975. Mutation selection and the natural history of cancer. Nature 255, 197–200 10.1038/255197a0 (doi:10.1038/255197a0) [DOI] [PubMed] [Google Scholar]
- 26.Traulsen A, Pacheco JM, Dingli D. 2007. On the origin of multiple mutant clones in paroxysmal nocturnal globinuria. Stem Cells 25, 3081–3084 10.1634/stemcells.2007-0427 (doi:10.1634/stemcells.2007-0427) [DOI] [PubMed] [Google Scholar]
- 27.Dingli D, Pacheco JM, Traulsen A. 2008. Multiple mutant clones in blood rarely coexist. Phys. Rev. E 77, 021915. 10.1103/PhysRevE.77.021915 (doi:10.1103/PhysRevE.77.021915) [DOI] [PubMed] [Google Scholar]
- 28.Lenaerts T, Pacheco JM, Traulsen A, Dingli D. 2010. Tyrosine kinase inhibitor therapy can cure chronic myeloid leukemia without hitting leukemic stem cells. Haematologica 95, 900–907 10.3324/haematol.2009.015271 (doi:10.3324/haematol.2009.015271) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. 2007. Tumor growth need not be driven by rare cancer stem cells. Science 317, 337. 10.1126/science.1142596 (doi:10.1126/science.1142596) [DOI] [PubMed] [Google Scholar]
- 30.Adams JM, Strasser A. 2008. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 68, 4018–4021 10.1158/0008-5472.CAN-07-6334 (doi:10.1158/0008-5472.CAN-07-6334) [DOI] [PubMed] [Google Scholar]
- 31.Kimura M. 1955. Solution of a process of random genetic drift with a continuous model. Proc. Natl Acad. Sci. USA 41, 144–150 10.1073/pnas.41.3.144 (doi:10.1073/pnas.41.3.144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waxman D. 2011. A compact result for the time-dependent probability of fixation at a neutral locus. J. Theor. Biol. 274, 131–135 10.1016/j.jtbi.2011.01.019 (doi:10.1016/j.jtbi.2011.01.019) [DOI] [PubMed] [Google Scholar]
- 33.Crow JF, Kimura M. 1970. An introduction to population genetics theory. New York, NY: Harper and Row [Google Scholar]
- 34.Ewens WJ. 2004. Mathematical population genetics. I. Theoretical introduction. New York, NY: Springer [Google Scholar]
- 35.Altrock PM, Traulsen A. 2009. Fixation times in evolutionary games under weak selection. New J. Phys. 11, 013012. 10.1088/1367-2630/11/1/013012 (doi:10.1088/1367-2630/11/1/013012) [DOI] [Google Scholar]
- 36.Parker C. et al. 2005. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 106, 3699–3709 10.1182/blood-2005-04-1717 (doi:10.1182/blood-2005-04-1717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murakami Y, Inoue N, Shichishima T, Ohta R, Noji H, Maeda Y, Nishimura J-I, Kanakura Y, Kinoshita T. 2012. Deregulated expression of HMGA2 is implicated in clonal expansion of PIGA deficient cells in paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 156, 383–387 10.1111/j.1365-2141.2011.08914.x (doi:10.1111/j.1365-2141.2011.08914.x) [DOI] [PubMed] [Google Scholar]
- 38.Luzzatto L. 1997. Somatic mutation in paroxysmal nocturnal hemoglobinuria. Hosp. Pract. (Minneap.) 32, 125–140 [DOI] [PubMed] [Google Scholar]
- 39.Rowley JD. 1973. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 243, 290–293 10.1038/243290a0 (doi:10.1038/243290a0) [DOI] [PubMed] [Google Scholar]
- 40.Bessler M, Mason P, Hillmen P, Luzzatto L. 1994. Somatic mutations and cellular selection in paroxysmal nocturnal haemoglobinuria. Lancet 343, 951–953 10.1016/S0140-6736(94)90068-X (doi:10.1016/S0140-6736(94)90068-X) [DOI] [PubMed] [Google Scholar]
- 41.Nafa K, Bessler M, Deeg HJ, Luzzatto L. 1998. New somatic mutation in the PIG-A gene emerges at relapse of paroxysmal nocturnal hemoglobinuria. Blood 92, 3422–3427 [PubMed] [Google Scholar]
- 42.Traulsen A, Pacheco JM, Luzzatto L, Dingli D. 2010. Somatic mutations and the hierarchy of hematopoiesis. BioEssays 32, 1003–1008 10.1002/bies.201000025 (doi:10.1002/bies.201000025) [DOI] [PubMed] [Google Scholar]
- 43.Marley SB, Lewis JL, Goldman JM, Gordon MY. 1998. Discordant erythropoiesis in CML. Leukemia 12, 444. 10.1038/sj.leu.2400955 (doi:10.1038/sj.leu.2400955) [DOI] [PubMed] [Google Scholar]
- 44.Nieborowska-Skorska M, et al. 2012. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 119, 4253–4263 10.1182/blood-2011-10-385658 (doi:10.1182/blood-2011-10-385658) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakraborty S, Stark JM, Sun CL, Modi H, Chen W, O'Connor TR, Forman SJ, Bhatia S, Bhatia R. 2012. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage–fusion–bridge cycles mediated by increased nonhomologous end joining. Blood 119, 6187–6197 10.1182/blood-2011-05-352252 (doi:10.1182/blood-2011-05-352252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao RC, Jiang Y, Verfaillie CM. 2001. A model of human p210(bcr/ABL)-mediated chronic myelogenous leukemia by transduction of primary normal human CD34(+) cells with a BCR/ABL-containing retroviral vector. Blood 97, 2406–2412 10.1182/blood.V97.8.2406 (doi:10.1182/blood.V97.8.2406) [DOI] [PubMed] [Google Scholar]
- 47.Sirard C. et al. 1996. Normal and leukemic SCID-repopulating cells (SRC) coexist in the bone marrow and peripheral blood from CML patients in chronic phase, whereas leukemic SRC are detected in blast crisis. Blood 87, 1539–1548 [PubMed] [Google Scholar]
- 48.Kelly R, Richards S, Hillmen P, Hill A. 2009. The pathophysiology of paroxysmal nocturnal hemoglobinuria and treatment with eculizumab. Ther. Clin. Risk Manag. 5, 911–921 10.2147/TCRM.S3334 (doi:10.2147/TCRM.S3334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alfinito F. et al. 2012. Eculizumab treatment modifies the immune profile of PNH patients. Immunobiology 217, 698–703 10.1016/j.imbio.2011.11.009 (doi:10.1016/j.imbio.2011.11.009) [DOI] [PubMed] [Google Scholar]
- 50.Stratton MR, Campbell PJ, Futreal PA. 2009. The cancer genome. Nature 458, 719–724 10.1038/nature07943 (doi:10.1038/nature07943) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jagers P. 1975. Branching processes with biological applications. London, UK: Wiley [Google Scholar]
- 52.Kimmel M, Axelrod DE. 2002. Branching processes in biology. New York, NY: Springer [Google Scholar]
- 53.Haccou P, Jagers P, Vatutin VA. 2005. Branching processes: variation, growth, and extinction of populations. Cambridge, UK: Cambridge University Press [Google Scholar]