

Abstract

Studies report a bidirectional SE' strategy applicable for the stereocontrolled synthesis of nonracemic 1,5-syn and 1,5-anti diols and their derivatives. Nonracemic 1,3,2-diazaborolidine auxiliaries are incorporated by chemoselective tin—boron exchange to provide reactive allylic boranes. The convergent pathway utilizes sequential reactions with two aldehydes producing stereochemical outcomes from cyclic, closed and open transition state preferences, respectively. Synthesis of fragment 16 of peloruside A is accomplished in four steps from readily available aldehydes 9 and 13.
Bifunctional reagents possessing properties of high chemoselectivity greatly facilitate convergent synthesis strategies. Several years ago, we initiated studies of bifunctional stannanes and silanes which provide for the sequential execution of SE' reactions and cross-coupling processes as an efficient strategy for the rapid assembly of molecular complexity.1 Efforts toward pelorusides A and B,2 have led us to expand the scope of these studies to consider bifunctional lynchpin species for sequential SE' operations and the stereocontrolled preparation of 1,5-syn and 1,5-anti diols. Currently, few reagents address this challenge as a versatile synthesis strategy affording high enantioselectivity.
Conceptually, a trimethylenemethane dianion equivalent may sequentially react with two different aldehydes for the construction of nonracemic 1 and 2 (Scheme 1). Our studies introduced the additional requirement to chemically distinguish the individual secondary alcohols for subsequent site-specific manipulations. A goal of these studies was to obtain nonracemic skipped polyol derivatives 3 and 4 via a mild oxidative cleavage of the central C=C followed by stereoselective hydride reduction.
Scheme 1.

General Plan for the Synthesis of Nonracemic Polyol Derivatives
Previous studies by Barrett and coworkers have described an asymmetric bidirectional double allylboration to yield C2-symmetric 3-methylenepentane-1,5-diols 1 (R1 = R2) using nonracemic 1,3-bis(diisopinocampheylboryl)-2-methylenepropanes.3 Roush and coworkers have explored the introduction of two different aldehydes in the double allylboration sequence of 1,3-diborylpropenes by taking advantage of reactivity differences of the borane moieties and by limiting the amount of the first aldehyde.4 Interestingly, Soderquist and coworkers have prepared borabicyclo[3.3.2]decane-derived 1,3-diborylpropenes which undergo 1,3-borotropic shifts to react as 1,1-bimetallic allylation species. Sequential reactions occur first with ketones, and subsequent additions to aldehydes produce 2-vinyl-1,3-diols of high enantiomeric purity.5
Properties of reactivity and selectivity of SE' reactions using silyl-substituted allylmetal reagents have primarily been investigated for examples leading to 1,2-anti-β-hydroxy allylsilanes.6 Peng and Hall have described a variation of this theme in which enantiocontrolled allylation results in the participation of an allylic silane in an intramolecular Prins reaction affording nonracemic 1,2,4-trisubstituted tetrahydrofurans.7 A significant precedent was established by Keck and coworkers using 2-(trimethylsilylmethyl)allyltri-n-butylstannane for the convergent union of two aldehydes in an asymmetric allylation-Prins sequence to yield 2,6-cis-disubstituted-4-methylenetetrahydropyrans.8
Our previous studies have utilized the mild and quantitative replacement of allylic stannanes with chiral, nonracemic boranes as an effective strategy for asymmetric allylation.9 This technique tolerates a wide range of functionality and has proven to be extremely useful in the synthesis of complex natural products.10 While the transmetallation of the stannane is highly favored, allylic trimethylsilanes were found to be inert. This observation established a basis for our studies of sequential allylation processes utilizing an initial reagent-controlled asymmetric reaction. These efforts have been applied for a stereocontrolled synthesis of the C(1)–C(9) fragment of peloruside A (5) as shown in Scheme 2. Thus, facile transmetallation of allylic stannane 611 using the bromoborane 712 provided in situ generation of nonracemic 1,3,2-diazaborolidine 8 upon stirring at 22 °C for 16 h. The (R,R)-1,2-diamino-1,2-diphenylethylene N,N-sulfonamide was incorporated as an effective chiral controller.13 A rapid SE' reaction was observed upon cooling to −78 °C with the addition of nonracemic aldehyde 9 (1.0 equiv)14 and 2,6-di-tert-butyl-4-methylpyridine (1.1 equiv)15 leading to stereoselective production of homoallylic alcohol 10 (80%).
Scheme 2.

Synthesis of the C1–C9 Fragment of Peloruside A (5)
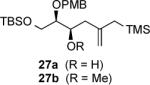
The outcome of the reaction is dominated by face selectivity imposed by the (R,R)-auxiliary. The closed, six-membered arrangement TS-1 leading to 10 also features the polar Felkin model for nucleophilic addition to the α-alkoxyaldehyde 9. Major product (R)-10 (R = H) (dr 89:11) was purified, and its stereochemistry was assigned by a modified Mosher ester analysis.16 O-Methylation and silyl ether formation gave 11 and 12, respectively, and the second stage of the process was undertaken as a Sakurai-Hosomi allylation. Precomplexation of α-alkoxyaldehyde 1317 with MgBr2 etherate in methylene chloride at −26 °C was followed by addition of silane 11 leading to selective formation of homoallylic alcohol 14 (80% yield; dr 25:1). An open, synclinal transition state features the chelation-controlled arrangement TS-2 to afford the major diastereomer. Reaction of the OTIPS derivative 12 provides slightly reduced yields and diastereoselectivity resulting in 15. In each case, the stereochemistry of the new alcohol (C3) has been established using the Mosher ester technique.16 Expedient construction of the C1–C9 polyketide fragments 16 and 17 is completed in excellent yield by the Johnson—Lemieux cleavage of the central (C5) alkene.
Strategically, the convergent pathway of Scheme 2 produces 1,5-anti diol derivatives 16 and 17 by using two chemodivergent SE' allylations. This characteristic is advantageous for planning syntheses of complex substances by sequential reactions employing closed and open transition state preferences. The potential and the generality of these processes have been explored. Table 1 offers a compilation of representative results for reactions of chiral, nonracemic and achiral aldehydes with (R,R)-7 and (S,S)-7 using the conditions developed in Scheme 2 as a standard protocol. We have noted that allylations with (R,R)- and (S,S)-diazaborolidines of 8 using enantiomeric aldehydes 18 and 19 express modest levels of matched and mismatched characteristics (entries 2 and 3), and the stereochemistry of the newly formed alcohol is consistently determined by the chiral controller. Alkyl branching at the α-position of nonracemic aldehydes 20 and 21 leads to transition states which provide for Felkin-Anh additions in entries 4 and 5. Yields are typically in the range of 79–83%. Beta-branching (entry 8) is less effective, and achiral aldehydes were included to provide a measure of asymmetric induction due to the allylborane reagent. Generally, the latter cases undergo the SE' reaction with approximately 90% face selectivity (entries 6 and 7). However, entries 6, 7 and 8 were not optimized in this study and led to the recovery of small quantities of starting aldehyde using the standard protocol. In practice, product purifications were conducted by flash chromatography using deactivated silica gel by the introduction of 0.5% NEt3 in the eluent to avoid protodesilylation. The stereoselectivity of each reaction was measured by HPLC analysis of the crude homoallylic alcohols to determine diastereomeric ratios (entries 1–5) or by direct conversion to their corresponding Mosher esters for integration of characteristic NMR signals (entries 6–8). In our initial studies, the purification of major products 26, 27, 28, and 32 (entries 2, 3, 4 and 8) by flash column chromatography was followed by O-methylation or formation of TBS silyl ethers. Subsequently, the crude alcohols were efficiently converted into their methyl ethers (entries 1, 5, 6 and 7) for flash chromatography immediately prior to the second SE' event. Methyl ethers are incorporated to accommodate efforts toward peloruside A (5) and to simplify our analyses of proton NMR spectra for subsequent products of Table 2. However, benzylic, TBS, and TIPS silyl ethers have also been utilized at this stage with little overall effect on the observed yields or diastereoselectivities.
Table 1.
Allylsilane Formation via SE′ Reactions of 1,3,2-Diazaborolidine Intermediatesa

| entry | aldehyde | borane 7 | major productd | yield (%) ratios [dr/er] |
|---|---|---|---|---|
| 1 |

|
(S,S) |

|
80 95:5b |
| 2 |

|
(R,R) |

|
83 98:2b |
| 3 |

|
(R,R) |
|
70 91:9b |
| 4 |

|
(S,S) |

|
81 98:2b |
| 5 |

|
(S,S) |

|
79 95:5b |
| 6 |
|
(R,R) |

|
60 85:15c |
| 7 |

|
(R,R) |

|
63 91:9c |
| 8 |

|
(R,R) |
|
58 91:9c |
Reaction conditions: Stannane 6 was reacted with bromoborane 7 in CH2C12 at 22 °C for 16 h under N2. The mixture was then cooled to −78 °C under N2 and a solution of aldehyde (1.0 equiv) containing 4-methyl-2,6-di-tert-butylpyridine was added via syringe. Reactions were stirred at −78 °C for 4 h and quenched by the addition of aqueous NaHCO3.
Ratios [dr] were were determined by HPLC analysis of the crude product mixture.
Ratios of enantiomers were determined by conversion to their corresponding Mosher esters followed by integration of NMR signals.
Major products were purified by chromatography using deactivated silica gel.
Table 2.
Second-stage Bidirectional SE′ Reactionsa

| entry | aldehyde | silane | major productd | yield (%) ratios [dr] |
|---|---|---|---|---|
| 1 |

|
26b (R = CH3) |

|
80 >20:1b |
| 2 |

|
26b (R = CH3) |

|
78 >20:1b |
| 3 |

|
25 (R = CH3) |

|
84 >20:1b |
| 4 |

|
25 (R = CH3) |

|
87 >20:1b |
| 5 |

|
25 (R = CH3) |

|
89 40:1c |
| 6 |

|
28b (R = CH3) |

|
80 >20:1b |
| 7 |

|
28b (R = CH3) |

|
84 40:1c |
| 8 |

|
29 (R = CH3) |

|
80 40:1c |
| 9 |

|
29 (R = CH3) |

|
82 >20:1b |
| 10 |

|
31 (R = TBS) |

|
75 >20:1b |
Reaction conditions: Aldehyde (1.1 equiv) was precomplexed with MgBr2•OEt2 (1.5 equiv) in CH2Cl2 at −78 °C under N2. A solution of allylsilane (1.0 equiv) was added in CH2Cl2 and the mixture was warmed to −26 °C with continuous stirring (12 h).
Ratios of diastereomers were determined for crude product mixtures by proton NMR analysis at 400 MHz.
Ratios of diastereomers were determined by analytical HPLC.
Major products were purified by flash chromatography and were fully characterized.
Table 2 summarizes results for second stage reactions with the stereocontrolled production of various acyclic polyol arrangements. In these examples, we have engineered high stereoselectivity for the formation of 1,5-syn- and 1,5-anti-products via the incorporation of nonracemic α-alkoxyaldehydes.18 Sakurai-Hosomi reactions proceed smoothly at −26°C in CH2Cl2 through open transition states via chelation-controlled additions to the less hindered face of precomplexed aldehydes. Yields ranging from 78–89% (dr >20:1) are routinely obtained. Significantly, the pre-existing chirality embedded within the allylsilane at the homoallylic site does not affect the stereoselectivity or the efficiency of the Sakurai reaction (compare entries 1 versus 2, and 3 versus 4; Table 2). In this fashion, the resulting 1,5-syn- and 1,5-anti-stereochemistry of the product is dictated by the choice of the nonracemic α-alkoxyaldehyde. The generality of the process also accommodates a number of common protecting groups (including PMB, MOM, MEM, TBS and related silyl ethers) which leads to a densely hydroxylated carbon framework in which each latent alcohol can be differentiated for further studies.
In summary, we have described a bidirectional SE' allylation strategy for stereoselective syntheses of acyclic 1,5-syn- or 1,5-anti-polyols. A chemoselective tin-boron exchange has allowed in situ production of chiral, nonracemic allylboranes containing an allylic silane. This feature facilitates a rational and efficient preparation of polyols via two orthogonal SE' processes. In this context, the boron reagent-controlled asymmetric allylation occurs via a preferred six-membered transition state and is followed by a Sakurai-Hosomi reaction demonstrating the stereochemical outcome of substrate control. Our results suggest broad applications toward the synthesis of stereochemically complex polyols.
Supplementary Material
Acknowledgment
The authors acknowledge financial support for this research, in part, from the National Institutes of Health (GM041560), National Science Foundation (CHE1055441), and Indiana University.
Footnotes
Supporting Information Available Experimental procedures; spectroscopic data; and selected 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Williams DR, Fultz MW. J. Am. Chem. Soc. 2005;127:14550–14551. doi: 10.1021/ja054201k. Williams DR, Morales-Ramos ÁI, Williams CM. Org. Lett. 2006;8:4393–4396. doi: 10.1021/ol0613160. Williams DR, Shah AA. Chem. Commun. 2010;46:4297–4299. doi: 10.1039/c0cc00679c. For use in total synthesis: Williams DR, Meyer KG. J. Am. Chem. Soc. 2001;123:765–766. doi: 10.1021/ja005644l.
- (2).(a) West LM, Northcote PT, Battershill CN. J. Org. Chem. 2000;65:445–449. doi: 10.1021/jo991296y. [DOI] [PubMed] [Google Scholar]; (b) Singh AJ, Xu C-X, Xu X, West LM, Wilmes A, Chan A, Hamel E, Miller JH, Northcote PT, Ghosh AK. J. Org. Chem. 2010;75:2–10. doi: 10.1021/jo9021265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barrett AGM, Braddock DC, de Koning PD, White AJP, Williams DJ. J. Org. Chem. 2000;65:375–380. doi: 10.1021/jo991205x. [DOI] [PubMed] [Google Scholar]
- (4).(a) Kister J, Nuhant P, Lira R, Sorg A, Roush WR. Org. Lett. 2011;13:1868–1871. doi: 10.1021/ol2003836. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flamme EM, Roush WR. J. Am. Chem. Soc. 2002;124:13644–13645. doi: 10.1021/ja028055j. [DOI] [PubMed] [Google Scholar]
- (5).González AZ, Román JG, Alicea E, Canales E, Soderquist JA. J. Am. Chem. Soc. 2009;131:1269–1273. doi: 10.1021/ja808360z. [DOI] [PubMed] [Google Scholar]
- (6).For a leading reference: Chen M, Roush WR. Org. Lett. 2011;13:1992–1995. doi: 10.1021/ol200392u.
- (7).Peng F, Hall DG. J. Am. Chem. Soc. 2007;129:3070–3071. doi: 10.1021/ja068985t. Sivasubramaniam U, Hall DG. Heterocycles. 2010;80:1449–1456. See also: Sarkar TK, Haque SA, Basak A. Angew. Chem. Int. Ed. 2004;43:1417–1419. doi: 10.1002/anie.200353184.
- (8).Keck GE, Covel JA, Schiff T, Yu T. Org. Lett. 2002;4:1189–1192. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Williams DR, Meyer KG, Shamim K, Patnaik S. Can. J. Chem. 2004;82:120–130. [Google Scholar]; (b) Williams DR, Kiryanov AA, Emde U, Clark MP, Berliner MA, Reeves JT. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12058–12063. doi: 10.1073/pnas.0402477101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Hennoxazole A, Williams DR, Brooks DA, Berliner MA. J. Am. Chem. Soc. 1999;121:4924–4925. [Google Scholar]; (b) Amphidinolide K, Williams DR, Meyer KG. J. Am. Chem. Soc. 2001;123:765–767. doi: 10.1021/ja005644l. [DOI] [PubMed] [Google Scholar]; (c) Leucascandrolide A, Williams DR, Plummer SV, Patnaik S. Tetrahedron. 2011;67:4949–4953. doi: 10.1016/j.tet.2011.05.020. See also: Williams, D. R.; Patnaik, S.; Plummer, S. V. Org. Lett. 2003, 5, 5035-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Phorboxazole A, Williams DR, Kiryanov AA, Emde U, Clark MP, Berliner MA. Angew. Chem. Int. Ed. 2003;42:1258–1262. doi: 10.1002/anie.200390322. [DOI] [PubMed] [Google Scholar]
- (11).For a preparation of stannane 6: Clive DLJ, Paul CC, Wang Z. J. Org. Chem. 1997;62:7028–7032. Also see Supporting Information
- (12).For information regarding the preparation of 7: (4R,5R)-2-Bromo-1,3-bis[(4-methylphenyl)sulfonyl]-4,5-diphenyl-1,3,2-diazaborolidine. e-EROS Encyclopedia of Reagents for Organic Synthesis [Online]; John Wiley & Sons, Ltd., Posted October 15, 2002. DOI: 10.1002/047084289X.rn00115.
- (13).(a) Corey EJ, Yu CM, Kim SS. J. Am. Chem. Soc. 1989;111:5495–5496. [Google Scholar]; (b) Williams DR, Brooks DA, Meyer KG, Clark MP. Tetrahedron Lett. 1998;39:7251–7254. [Google Scholar]
-
(14).Aldehyde 9 is readily prepared in four steps from (S)-(−)-glycidol (Aldrich) as illustrated below:

- (15).The addition of 2,6-di-tert-butyl-4-methylpyridine serves as an effective proton scavenger and suppresses side products of protodesilylation.
- (16).(a) Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512–519. [Google Scholar]; (b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092–4096. [Google Scholar]
-
(17).Nonracemic aldehyde 13 was prepared as shown below in three steps from dimethyl-L-tartrate. See: Williams DR, Klingler FD. Tetrahedron Lett. 1987;28:869–872..

- (18).For the preparation of nonracemic aldehyde 33: Corey EJ, Hannon FJ, Boaz NW. Tetrahedron. 1989;45:545–555.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.