

Abstract
Low-amplitude electric field (EF) is an important component of wound-healing response and can promote vascular tissue repair; however, the mechanisms of action on endothelium remain unclear. We hypothesized that physiological amplitude EF regulates angiogenic response of microvascular endothelial cells via activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway. A custom set-up allowed non-thermal application of EF of high (7.5 GHz) and low (60 Hz) frequency. Cell responses following up to 24 h of EF exposure, including proliferation and apoptosis, capillary morphogenesis, vascular endothelial growth factor (VEGF) expression and MAPK pathways activation were quantified. A db/db mouse model of diabetic wound healing was used for in vivo validation. High-frequency EF enhanced capillary morphogenesis, VEGF release, MEK-cRaf complex formation, MEK and ERK phosphorylation, whereas no MAPK/JNK and MAPK/p38 pathways activation was observed. The endothelial response to EF did not require VEGF binding to VEGFR2 receptor. EF-induced MEK phosphorylation was reversed in the presence of MEK and Ca2+ inhibitors, reduced by endothelial nitric oxide synthase inhibition, and did not depend on PI3K pathway activation. The results provide evidence for a novel intracellular mechanism for EF regulation of endothelial angiogenic response via frequency-sensitive MAPK/ERK pathway activation, with important implications for EF-based therapies for vascular tissue regeneration.
Keywords: endothelial cells, angiogenesis, electric field, MAPK, ERK
1. Introduction
Endogenous physiological (40–250 mV mm−1) electric field (EF) is an important component of the body's wound-healing response [1]. Different types of low, physiological amplitude electromagnetic field have been shown to influence a wide variety of biological systems [2] and have been used as a therapeutic tool for tissue repair, including bone healing, soft tissue repair and the healing of chronic wounds [3–7]. However, the widespread acceptance of EF therapies for wound healing has been prevented by the lack of standardized protocols and associated variability in the healing outcomes [8]. This variability often stems from the arbitrary choice of EF therapeutic parameters [9], resulting from an incomplete understanding of the fundamental pathways that are involved in EF interactions with specific tissues.
There has been emerging evidence that certain types of EFs can promote blood vessel formation (angiogenesis) and tissue vascularization [10]. However, research focused on the angiogenic effects of EF has been limited to a small number of EF modalities [2], and no comprehensive relationship exists between EF orientation, frequency, amplitude and endothelial cell angiogenic responses. The process of angiogenesis includes endothelial cell activation by angiogenic factors or changes in the extracellular environment, followed by cell migration, proliferation, formation of nascent capillaries, vasculature remodelling and maturation [11]. Pulsed electric and magnetic field stimulation have been shown to enhance in vivo angiogenesis in both ischaemic and non-ischaemic rat limbs [5,12,13] and in mouse wound healing [14]. Migration, tubular formation, proliferation and vascular endothelial growth factor (VEGF) expression in human umbilical cord endothelial cells (HUVECs) were stimulated by direct current (DC) as well as pulsed electromagnetic fields [10,15–17].
Importantly, the majority of previous studies have used in-plane DC field configuration, where exposure to the DC EF resulted in dramatic cell reorientation and directional migration (electrotaxis) [10], as well as an altered pattern of integrin receptor clustering and the associated actin reorganization in endothelial cells and fibroblasts [2,18,19]. However, there is significant variability in EF-induced cell migration, not only between cells of different types [2], but also between endothelial cells of different origin. Thus, bovine aortic endothelial cells migrate towards cathode [18], while HUVECs migrate towards anode [10]. Overall, experimental evidence suggests that the mechanisms responsible for EF-mediated angiogenic endothelial cell activation may be different from those that govern electrotaxis. Therefore, stimulation of electrotaxis alone may not necessarily result in an overall enhanced angiogenic response and improved wound healing. This is consistent with the results of the clinical studies that suggest that a pulsed (not DC) EF may be the most efficient modality in the treatment of chronic wounds [7,8,20] and in alleviating the symptoms of multiple sclerosis [9,21,22]. Importantly, mechanistic understanding of EF effects on endothelial cells is essential for the informed choice of the field parameters for wound-healing therapies.
Among the intracellular responses that may be mediated by EF, mitogen-activated protein kinase (MAPK) signalling cascade family [23] is the primary candidate. Of this family, extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK) and stress-activated protein kinase-2 (p38) pathways are known to be involved in angiogenic as well as stress-activated signalling in the absence of EF [24–30]. There is also evidence that these pathways can be activated in response to EF. It has been reported that 900 MHz mobile phone radiation activated the heat shock protein 27 (Hsp27)/p38MAPK stress response pathway in human endothelial cells [31], while a 50 Hz sinusoidal magnetic field affected the cellular distribution of Hsp27 and increased Hsp70, but not Hsp27 mRNA in aortic endothelial cells [32]. Also, DC EF activated ERK, JNK and p38 in embryonic stem cells and induced endothelial differentiation [33]. Different types of electromagnetic fields have been shown to affect the activation of ERK, JNK and p38 in several non-endothelial cell types [34–36]. However, the role of different EF modalities on MAPK activation in endothelial cells is not understood.
Previous studies have shown that EF-induced intracellular responses in non-endothelial cells may depend on the field frequency [2,37]; however, the possible role of this parameter in angiogenic responses of endothelial cells to EF is not known. It has been suggested [38] that at frequencies below 100 MHz, the cell (including cytoplasm and nucleus) can be considered as a conductive media surrounded by high capacitance membrane, which results in excluding the field from the cell cytoplasm. In contrast, at higher frequencies (gigahertz range), the low membrane impedance allows the current to flow through intracellular space (dielectric behaviour), which results in the field penetration across the membrane. The experimental evidence in this area remains limited.
The objective of this study was to elucidate the possible intracellular mechanisms for EF-mediated angiogenic responses in endothelial cells in a controlled setting in the absence of electrotaxis, to allow direct mechanistic interpretation of the data. We tested the hypothesis that EF with amplitudes in the physiological range regulates endothelial angiogenic response via activation of MAPK/ERK pathway. Experiments were conducted by using a custom-engineered multi-component system for microvascular endothelial cell exposure to EF with spatially controlled field distribution, combined with cell culture, microscopy and molecular biology methods. Initial in vivo validation was performed using an in vivo db/db mouse model of diabetic wound healing.
2. Material and methods
2.1. Microvascular endothelial cell isolation and culture
Murine microvascular endothelial cells were isolated from the lungs of C57 mice (Jackson Laboratory, ME, USA), as described previously [39]. Cells were doubly sorted using PECAM-1- and ICAM-2-conjugated magnetic beads (Invitrogen Corporation, CA, USA) and cultured in medium M199 (HyClone, UT, USA) supplemented with 10 per cent foetal bovine serum (FBS; Atlanta Biologicals, GA, USA), 1 per cent antibiotic/antimycotic (AB/AM; Atlanta Biologicals), 1 per cent heparin (Sigma-Aldrich, MO, USA) and 10 ng ml−1 endothelial growth factor supplement (Sigma-Aldrich). Cells from passages four to nine were used. All experiments were conducted in the culture medium (medium M199, 10% FBS, 1% antibiotic/antimycotic and 1% heparin) without additional growth factor supplementation.
2.2. In vitro electric field exposure set-up
A high-frequency and a low-frequency set-up were built to allow cell exposure to EF with a well-characterized field distribution, which was confirmed for each frequency by numerical simulations, as described below.
2.2.1. High-frequency electric field set-up
A custom set-up was built that allowed EF exposure operating at 7.5 GHz frequency (figure 1a). This frequency represents the regime where the membrane impedance becomes low (dielectric behaviour), resulting in the field penetration across the membrane [38]. The high-frequency EF set-up operated in transverse magnetic mode (TM010), where the dominant EF was normal to the plane of the cultured cells, and the magnetic field at the location of the cells was approximately zero. The apparatus consists of a cylindrical cavity resonator made from a copper waveguide with length of 31.9 mm and diameter of 31.7 mm. The cavity resonator was placed in a temperature-controlled 5 per cent CO2 cell culture incubator and connected to a semi-rigid coax (Microcoax, PA, USA) transmission line supplying 7.5 GHz EF from a vector network analyser (Anritsu, CA, USA). Cells were seeded in 12 mm diameter culture insert (Millipore, MA, USA), which was placed in a small plastic dish filled with the culture medium (20 mm in diameter) located inside the cavity resonator. This dish was connected to a large reservoir outside the resonator to ensure a constant medium level. Once coupled, a frequency sweep of the reflected power showed a dip that occurred when the frequency matched the resonant frequency of the cavity (7.5 GHz). Under these critical coupling conditions, the reflected signal on resonance dropped, and more than 90 per cent power supplied by the coaxial was used to support the oscillating cavity mode (TM010). The quality factor (Q) of the cavity was 170, and the calculated field intensity for the set-up with the insert without cells was 156 mV mm−1, which is in physiological range [1].
Figure 1.

Experimental set-up for microvascular endothelial cells exposure to EF. (a) High-frequency (7.5 GHz) EF set-up: an insert with endothelial cells was placed in the small plastic dish inside the cavity resonator to which EF was delivered from a vector network analyser (VNA) via a coaxial line. Cell culture medium flowed continuously between the cell insert and reservoir outside the resonator. (b) Low-frequency (60 Hz) EF set-up: plastic dish with the cell insert was placed between the plates of a parallel-plate capacitor. Red arrows in (a) and yellow arrows in (b) indicate direction of the electric field component, which was perpendicular to the cell surface in both configurations. Both set-ups operated inside a cell culture incubator (37°C, 5% CO2). (c,d) Numerical calculation of the EF distribution using a three-dimensional model of the apparatus and ANSYS HFSS software demonstrated that while the EF distributions outside the insert were different between two configurations, EF distributions at the location of the cells were similar (c) and uniform in the central part of the insert (d). (e) Temperature of the sample medium remained constant at 37°C during EF exposure.
2.2.2. Low-frequency electric field set-up
The custom-built set-up used for low-frequency (60 Hz) EF exposure (figure 1b) consisted of a parallel-plate capacitor (135 × 128, 26 mm apart) assembled in the same cell culture incubator. This frequency is within the range where intracellular space is shielded by the applied field [38], and where the angiogenic effects of EF have been previously observed [5,12]. The plates of the capacitor were connected to an Agilent 33250A function/arbitrary waveform generator (Agilent Technologies, Inc., CA, USA) and an oscilloscope (Tektronix Inc., OR, USA). Endothelial cells were seeded in the culture insert, which was placed in a small dish located between the plates. The EF was normal to the cell plane, and the calculated field intensity for the set-up with the insert without cells was within physiological range (209 mV mm−1).
2.2.3. Numerical calculation of the electric field distribution
A detailed numeric calculation of the EF distribution in the high-frequency resonator and the low-frequency capacitor was performed using an accurate three-dimensional model of the apparatus and the sample insert using the ANSYS HFSS package (ANSYS, PA, USA). The simulation program calculated a solution of the Maxwell equations, using measured dimensions of the dielectric insert and the media container and inputs (microwave power and the capacitor excitation voltage). Independence of the software output of the grid density was ensured by performing multiple calculations on grids of different sizes and densities. Results demonstrated that the EF distributions at the location of the cells were within the physiological range and similar for both set-ups (figure 1c), as well as uniform in the central part of the insert (figure 1d). The upper bound for the field power-specific absorption rate (SAR) for the sample in the resonator was estimated for the assumption that all of the field power fed to the resonator (50 µW in all experiments reported here) is absorbed in the sample. Under these ‘worst case’ assumptions that significantly overestimate the absorption rate, the SAR value is at most 0.1 W kg−1, which is considerably lower than the SAR human health safety limit [40].
2.2.4. Temperature measurements
For temperature measurements, EF exposure was briefly stopped, and the recordings of the temperature in the culture medium were made using an infrared thermometer (Braun, OH, USA, 0.2°C accuracy) without taking the samples out of the exposure set-up. To confirm the accuracy of the temperature measurements, the following controls were included. First, a control sample was placed inside the same incubator as the EF exposure apparatus, but was not subjected to EF stimulation. Second, the temperature of a large medium reservoir located in the same incubator was measured. All measurements were performed in duplicates and experiments were repeated three times. The results show (figure 1e) that the average temperature of all samples did not change during EF exposure and remained within a 37 ± 0.12 degree interval with 95% confidence.
2.3. In vitro electric field experiments
Capillary morphogenesis and MAPK pathways activation were quantified following 12 h of EF exposure, and the analyses of VEGF expression, cell proliferation and apoptosis were conducted up to 24 h of EF exposure. The experimental groups included endothelial cells exposed to high-frequency EF, low-frequency EF and a group not exposed to EF.
2.4. In vitro capillary morphogenesis
Capillary morphogenesis was assessed using a nanofibre-based angiogenesis assay previously developed in the laboratory [41–43], in which endothelial cells seeded on RAD16-II peptide nanofibre hydrogel (RARADADARARADADA; SynBioSci Corporation, CA, USA) undergo spontaneous capillary morphogenesis with clearly identifiable lumens in the absence of external angiogenic growth factors. Endothelial cells were seeded on the surface of 1 per cent (w/v) hydrogel in cell culture inserts (Millipore) at a seeding density of 105 cells cm−2. Cells seeded on 5 per cent gelatin-coated inserts were used as a negative control. Cells were labelled with CellTracker dye (Invitrogen) before seeding or with Phalloidin-TRITC (Sigma-Aldrich). After EF exposure, samples (at least n = 10 separate EF exposure experiments per group) were fixed with 2 per cent formaldehyde, and images of the sample surface (n = 5 per sample) were captured at 20× magnification using an inverted fluorescent microscope (Olympus IX81; Olympus, PA, USA). The characteristic size of capillary-like networks was determined using correlation analysis and custom-written Matlab code (The Math Works, MA, USA) [43].
2.5. Cell proliferation and apoptosis
Cells were seeded (2 × 104 cells cm−2) on 5 per cent gelatin-coated culture inserts (Millipore). Some samples were incubated with bromodeoxyuridine (BrdU; Invitrogen) for 8 h prior to experiments. After 12 and 24 h of EF exposure, cells were immediately fixed (2% formaldehyde) and stained with either anti-BrdU antibody (Invitrogen) or with anti-active Caspase-3 antibody (Promega, WI, USA) followed by goat anti-rabbit Alexa Fluor 594 and 4′,6-diamidino-2-phenylindole nuclear staining (both from Invitrogen) to identify proliferating and apoptotic cells, respectively. Percentages of proliferating or apoptotic cells were determined from five images at 20× magnification per sample. For each assay, experiments were repeated four times.
2.6. Vascular endothelial growth factor and placenta growth factor protein expression
VEGF and placenta growth factor (PlGF) are two major angiogenic cytokines acting through VEGF receptors pathway. VEGF binds to both VEGFR1 and VEGFR2 receptors, although it signals through VEGFR2. By contrast, PlGF only binds to and signals through VEGFR1 [44,45]. To determine the effect of EF on the VEGF and PlGF protein release by endothelial cells, culture medium samples (at least n = 6 separate experiments) were used to measure VEGF and PlGF protein levels using appropriate ELISA kits (R&D Systems, MN, USA).
2.7. ERK, JNK, p38 MAPK pathways activation
After EF exposure, cells were lysed using buffer containing 20 mM Tris–HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 per cent Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg ml−1 leupeptin and 1 mM phenylmethylsulphonyl fluoride. The total protein concentration in each sample lysate was determined using Coomassie plus assay kit (Thermo Fisher Scientific, IL, USA). A 10 µg bolus of total protein was used for all MAPK pathway enzyme-linked immunosorbent assays (ELISAs). The total and phosphorylated levels of ERK, MEK, p38 and JNK proteins were quantified using appropriate sandwich ELISA kits (Cell Signaling Technology, MA, USA). MEK-cRaf complex levels and free (unbound) MEK levels were quantified according to previously described protocols with modifications (see the electronic supplementary material, figure S1) [46–48]. The total MEK levels are presented in optical densities and phosphorylated MEK, MEK-cRaf and pMEK-cRaf levels are normalized to total MEK levels. For negative control, β-actin (Invitrogen) was immunoprecipitated from the lysate and subjected to ELISAs. All ELISA assays were performed in duplicates or triplicates, with all experiments repeated at least six times.
2.8. Inhibitor studies
To determine the role of VEGF signalling in EF-mediated angiogenic responses, experiments were repeated in the presence of 0.1 µg ml−1 soluble anti-mouse VEGF blocking antibody (R&D Systems), or 5 µM SU5416 (Sigma-Aldrich), a specific pharmacological VEGFR2 inhibitor [49–51], or in the presence of 10 µM U0126 MEK inhibitor (Sigma-Aldrich). The efficiency of SU5416 has been verified using a standard approach of inducing MEK activation by 20 ng ml−1 VEGF and then inhibiting the response with SU5416 (see the electronic supplementary material, figure S2). To examine the role of PI3K, Ca2+ and endothelial nitric oxide synthase (eNOS) signalling in EF-mediated MEK activation, cells were treated with 10 µM LY294002 (PI3K inhibitor) [52], 10 µM 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) (Ca2+ chelator) [53] or 200 µM Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME, eNOS inhibitor) [54], respectively, and the total and phosphorylated MEK levels were measured using ELISA. All inhibitors were added to the culture supernatant and pre-incubated for 1 h to equilibrate respective target blocking prior to EF exposure. All analyses were done in duplicate/triplicate, and all inhibitor experiments were repeated at least three times.
2.9. In vivo diabetic wound-healing model and electric field treatment
Eight to 10 week old female BKS.Cg - m+/+Leprdb/J (db/db) mice with serum glucose levels greater than 450 mg dl−1 were used. Previous studies have shown that this animal model is characterized by a delayed wound healing, with reduced neovascularization of the repair tissue [41,55]. Two full-thickness excisional skin wounds (8 mm) were created on the back of the mice, washed with 50 µl of sterile phosphate-buffered saline and covered with sterile adhesive dressing (TegadermTM, 3 M, MN, USA) [55]. EF treatment of the wounds was achieved through a custom-built EF exposure set-up (see figure 8a), which included two antennae connected to the EF source (8350B Sweep Oscillator, Agilent Technologies) through a flexible co-axial cable. Prior to exposure, the animals were anaesthetized, and EF antennas were placed approximately 5 mm away from the wounds. EF stimulation of 7.5 GHz and approximately 200 mV mm−1 was applied for 1 h every day for 7 days. Control group included animals that underwent the same wounding procedure, but were not exposed to EF (n = 5 animals per group). All animals were sacrificed and wounds were harvested at day 8.
2.10. Vascular endothelial growth factor expression in the wounds
The harvested wounds were homogenized in 50 mM Tris–HCl buffer containing 1 per cent NP40, aprotinin (3.3 μg ml−1), leupeptin (10 μg ml−1) and pepstatin (4 μg ml−1). VEGF protein expression in wound tissue homogenate was measured using ELISA kit (R&D Systems).
2.11. Statistical analyses
The results are reported as average ± s.d. Multi-factor ANOVA and post hoc tests with Bonferroni corrections (SPSS, IL, USA) were used to test for the effects of EF, field frequency and the inhibitors on the capillary morphogenesis, VEGF expression and total as well as phosphorylated levels of MAPK pathway proteins. Results were considered statistically significant at p < 0.05.
3. Results
3.1. Electric field enhances angiogenic response by microvascular endothelial cells
In the capillary morphogenesis assay used in this study, endothelial cells undergo spontaneous formation of multi-cellular capillary structures with clearly identifiable lumens by 12 h of cell seeding on the nanofibre hydrogel [41,42]. High-frequency EF exposure resulted in significantly larger structures, when compared with low-frequency and no-EF groups (p < 0.001, figure 2a,b), while no significant differences between low-frequency and no-EF groups were observed. Similarly, VEGF expression was significantly increased in cells exposed to high-frequency EF, when compared with low-frequency or no-EF groups (p < 0.001, figure 2c), while there was no significant difference in VEGF levels between low-frequency and no-EF groups. The pro-angiogenic effects of EF were not associated with EF-induced directional cell responses (electrotaxis), as demonstrated by the absence of cell re-orientation when seeded on the gelatin-coated inserts in this field configuration (see the electronic supplementary material, figure S3). Interestingly, the effects of EF on both capillary morphogenesis and VEGF released by endothelial cells were retained in the presence of soluble anti-VEGF blocking antibody (p < 0.05, figure 3a), when compared with the no-EF group. An addition of potent VEGFR2 receptor inhibitor SU5416 completely abolished capillary morphogenesis and significantly reduced VEGF release in all experimental groups, including no-EF controls (figure 3b). Next, we investigated the effects of EF on the signalling downstream of VEGFR2 by quantifying the phosphorylation of MEK, which is upstream of ERK. High-frequency EF resulted in increased MEK phosphorylation, where the magnitude of the effect did not depend on the presence of SU5416 or exogenous VEGF and remained at the 1.5-fold to twofold levels (figure 3c). This effect was not present in the low-frequency group. These results suggest that external VEGF binding to its receptor may not be required for pro-angiogenic effects of EF in this system, and that the EF stimulation is not strong enough to reverse a complete inhibition of VEGFR2-mediated angiogenesis induced by SU5416. However, the EF-induced VEGFR2-independent activation of the MEK/ERK pathway may be responsible for the increased release of VEGF observed in the high-frequency group (figure 2c) and potentially activation of the VEGF autocrine loop.
Figure 2.

Electric field enhances angiogenic response by microvascular endothelial cells. (a) Cell exposure to high-frequency EF significantly enhanced capillary morphogenesis, with clear multi-cellular tubule formation (20× magnified image, green: endothelial cells, blue: cell nuclei), resulting in larger characteristic network size (b) when compared with low-frequency and no-EF groups (n = 10, p < 0.001). Scale bar, 100 µm. (c) High-frequency EF stimulation resulted in significantly higher levels of VEGF released into the culture medium, when compared with the no-EF group (n = 3, p < 0.01).
Figure 3.

Angiogenic effects of EF do not require VEGF binding to VEGFR2 receptor. The effects of EF on capillary morphogenesis and VEGF release into the medium were retained in the presence of VEGFR2 blocking antibody (n = 4, p < 0.05), suggesting that EF-mediated stimulation of angiogenesis does not require VEGF ligand–receptor binding (a). Treatment of endothelial cells with a potent VEGFR2 inhibitor SU5416 effectively abolished capillary morphogenesis and VEGF release (b), but not high-frequency EF-induced MEK phosphorylation, which was significantly higher in the high-frequency group even in the presence of SU5416, when compared with low-frequency and no-EF controls (c). Interestingly, the relative magnitude of high-frequency EF-induced MEK phosphorylation normalized to no-EF (0.41 ± 0.09), no-EF + SU5416 (0.25 ± 0.04), no-EF + SU5426 + VEGF (0.44 ± 0.30), respectively, did not depend on the presence of SU5416 or exogenous VEGF and remained 1.5- to twofold higher than no-EF levels (p < 0.05, n = 3). This effect was not present in the low-frequency group.
To further confirm the involvement of the MEK/ERK pathway in EF-mediated angiogenic responses, capillary morphogenesis and VEGF release by endothelial cells were quantified in the presence of high-affinity MEK inhibitor U0126. These responses were significantly reduced in all experimental groups, when compared with no inhibitor controls (p < 0.05, figure 4a,b). Interestingly, treatment with U0126 effectively reversed the effect of high-frequency EF on capillary morphogenesis and VEGF release, where significantly lower values for characteristic network size and VEGF release were observed, when compared with those in low-frequency EF and no-EF groups (p < 0.001). There was no significant difference in network size between low-frequency EF and no-EF groups in the presence of U0126. There was no effect of EF exposure on PlGF release by endothelial cells when compared with the no-EF group (see the electronic supplementary material, figure S3).
Figure 4.
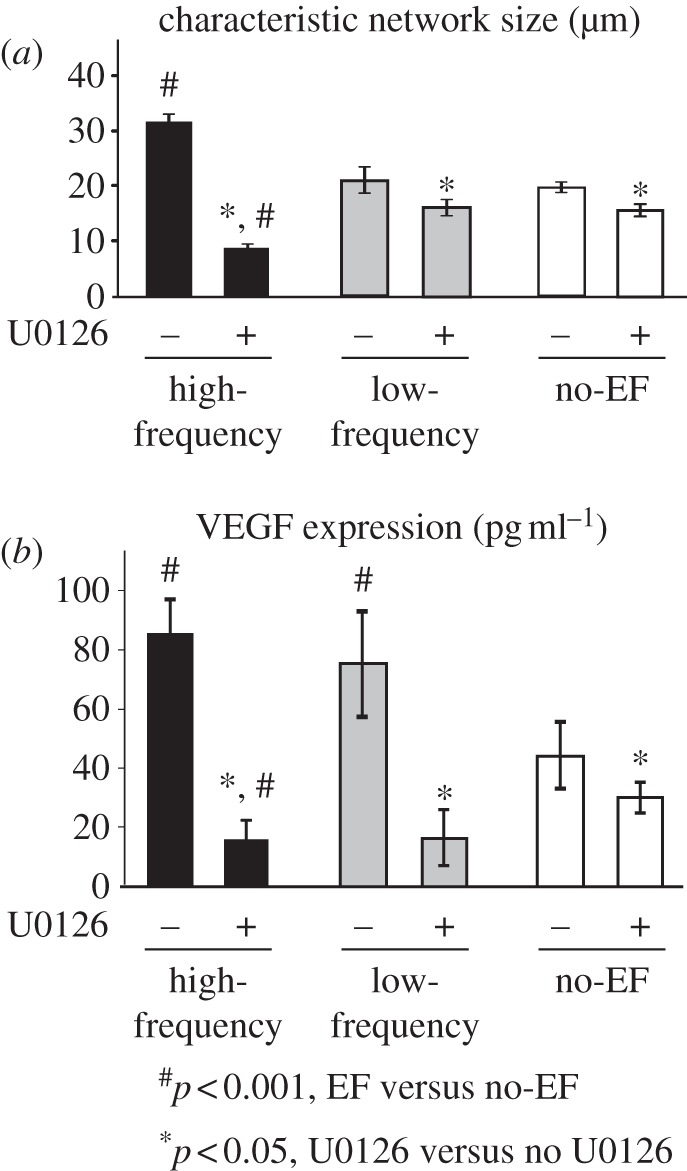
MAPK/ERK pathway is involved in EF-mediated angiogenic response. (a) Treatment with MEK inhibitor U0126 resulted in decreased characteristic network size in all groups (n = 5, p < 0.05), and effectively abolished the increase in capillary morphogenesis in the high-frequency EF group, when compared with low-frequency EF and no-EF groups (n = 5, p < 0.001), suggesting that angiogenic effects of EF involve the MEK/ERK pathway. (b) Similarly, the VEGF release by endothelial cells in the presence of U0126 was significantly reduced in all groups, with VEGF levels significantly smaller in high-frequency EF than no-EF group (n = 4, p < 0.001).
3.2. High-frequency electric field increases ERK, but not JNK or p38 phosphorylation in endothelial cells
Both high-frequency and low-frequency EF did not affect the total levels of ERK, JNK or p38 protein expression (figure 5a). However, cell exposure to high-frequency EF resulted in significantly increased levels of ERK phosphorylation, when compared with the no-EF group (p < 0.001, figure 5b), while no significant effects of high-frequency EF on JNK and p38 phosphorylation were observed. Also, endothelial cells exposed to low-frequency EF had significantly lower levels of ERK and p38 phosphorylation, when compared with no-EF group, while no differences were detected in phosphorylated JNK levels between low-frequency EF and no-EF groups (p < 0.001, figure 4b).
Figure 5.
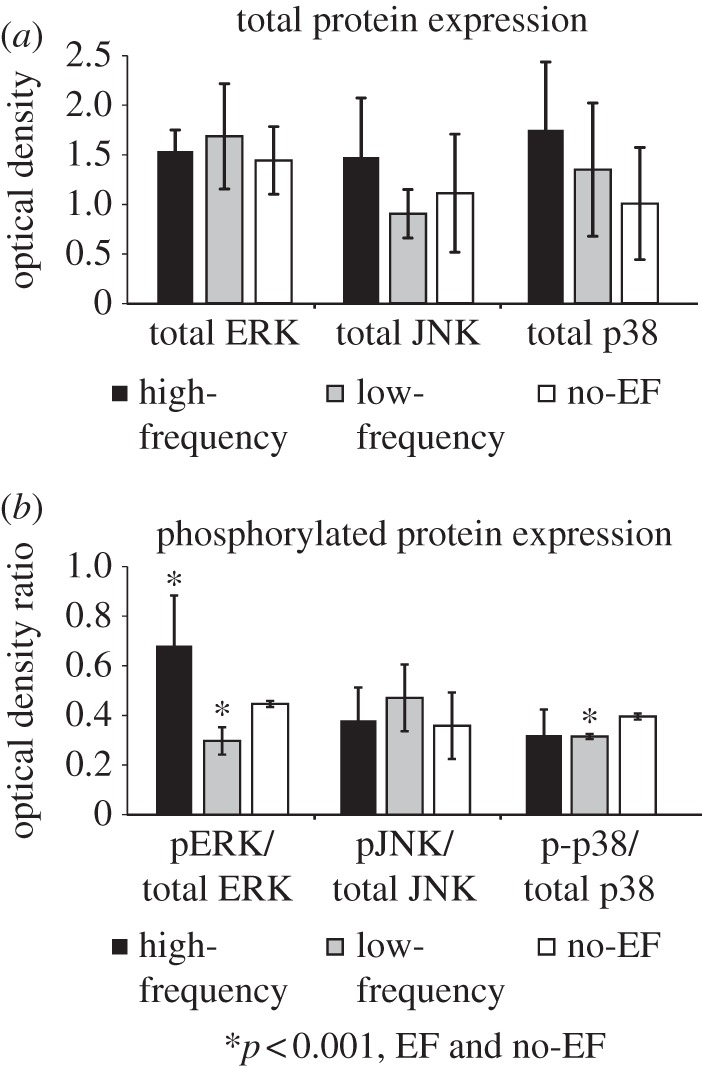
High-frequency EF increases the phosphorylation of ERK, but not JNK or p38 MAP kinase in microvascular endothelial cells. (a) Exposure to EF did not alter the total protein levels of ERK, JNK or p38. (b) High-frequency EF significantly increased phosphorylation of ERK, when compared with low-frequency or no-EF groups (n = 6, p < 0.001). In contrast, no effect of high-frequency EF on JNK or p38 phosphorylation relative to no-EF controls was observed. In the low-frequency EF group, phosphorylated levels of ERK and p38 were significantly decreased when compared with the no-EF group (n = 6, p < 0.001).
3.3. High-frequency electric field enhances MEK phosphorylation and MEK-cRaf complex formation in endothelial cells
Cell exposure to high-frequency EF resulted in significantly higher levels of phosphorylated MEK, while the total MEK levels remained unchanged with EF exposure both in the absence or the presence of MEK inhibitor U0126 (figure 6a). Previous studies have shown that the binding of upstream cRaf with MEK at serine 218 and serine 222 motifs [56] is necessary for MEK phosphorylation and downstream pathway activation. Therefore, to determine the involvement of this MAPK/MEK-ERK pathway in EF-induced angiogenic affects, the levels of cRaf bound to MEK (MEK-cRaf complex), levels of MEK phosphorylation within the complex (pMEK-cRaf) and the free MEK (unbound MEK) levels were quantified. Results showed that cell exposure to high-frequency EF significantly increased protein levels and phosphorylation of the MEK–cRaf complex, when compared with low-frequency or no-EF groups (figure 6b, p < 0.001). This was consistent with low levels of free (unbound) MEK in the high-frequency EF group, when compared with those in low-frequency and no-EF groups (figure 6b, p < 0.05). In the case of low-frequency EF exposure, the free MEK levels were significantly higher than high-frequency and no-EF groups (p < 0.05, figure 6b). In contrast to the EF effects observed in the absence of U0126 (figure 6b), cell exposure to high-frequency EF in the presence of U0126 resulted in reduced protein levels and phosphorylation of the MEK–cRaf complex, as well as increased free MEK levels (figure 6c, p < 0.05), when compared with low-frequency or no-EF groups (n = 7, p < 0.01).
Figure 6.

High-frequency EF enhances MEK phosphorylation and MEK–cRaf complex formation in microvascular endothelial cells. (a) Cell exposure to high-frequency EF resulted in significantly higher protein levels of phosphorylated MEK, while the total levels of MEK remained unchanged with EF exposure both in the absence or presence of MEK inhibitor U0126. (b) Cell exposure to high-frequency EF significantly increased protein levels and phosphorylation of MEK-cRaf complex, when compared with low-frequency or no-EF groups (n = 7, p < 0.001). This was consistent with low levels of free (unbound) MEK in the high-frequency EF group, when compared with those in low-frequency and no-EF groups (n = 7, p < 0.05). Free MEK levels in the low-frequency group were significantly larger than the values in the high-frequency and no-EF groups (n = 7, p < 0.05). (c) In contrast to the EF effects observed in the absence of U0126 in (b), cell exposure to high-frequency EF in the presence of MEK inhibitor resulted in significantly reduced protein levels and phosphorylation of MEK-cRaf complex, as well as increased free MEK levels (n = 7, p < 0.05), when compared with low-frequency or no-EF groups (n = 7, p < 0.01).
3.4. Effects of PI3K, eNOS inhibition and Ca2+ chelation on electric field-mediated MEK phosphorylation in endothelial cells
PI3K is another upstream mediator of the MAPK/MEK pathway, in addition to VEGFR2 [54,57]. In our experiments, inhibition of PI3K resulted in a significant reduction in MEK phosphorylation, as expected based on the previous reports [49,58]. However, the stimulatory effect of high-frequency EF on MEK phosphorylation was still retained even in the presence of LY294002 (PI3K inhibitor), where pMEK levels were significantly greater in EF groups, when compared with no-EF controls (p < 0.05, figure 7a).
Figure 7.
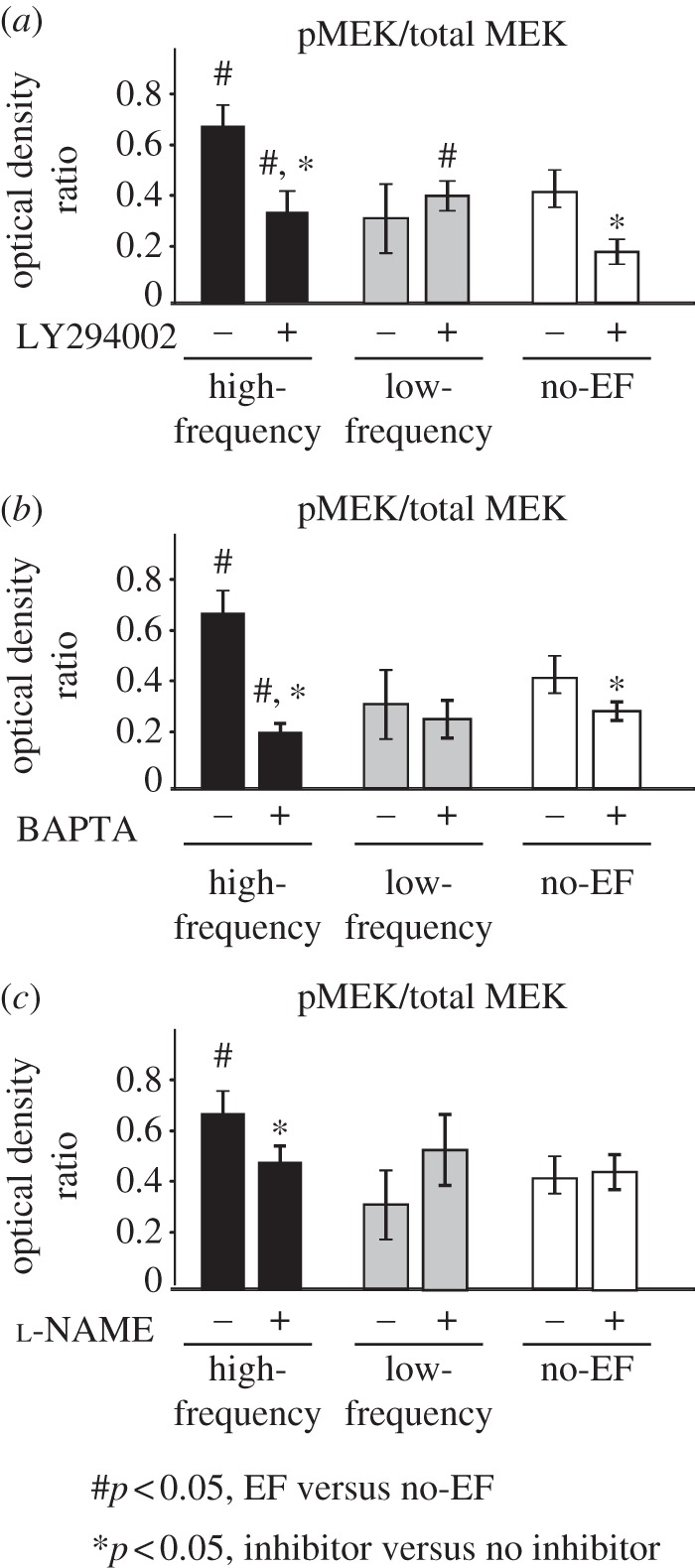
Effects of PI3K and eNOS inhibition and Ca2+ chelation on EF-mediated MEK phosphorylation: (a) addition of PI3K inhibitor LY294002 did not abolish EF-induced increase in pMEK levels, when compared with no-EF controls (n = 4, p < 0.05), suggesting that this pathway may not be critical for EF-mediated angiogenic cell responses. (b) Interestingly, addition of Ca2+ chelator BAPTA resulted in significantly reduced pMEK levels, when compared with no-EF controls, which was similar to the trends in cell responses observed in the presence of MEK inhibitor (figure 4). (c) eNOS inhibition using l-NAME did not affect pMEK levels in low-frequency and no-EF groups, and effectively abolished high-frequency EF-induced increase in MEK phosphorylation (n = 4, p < 0.05). These results indicate the involvement of Ca2+ and eNOS pathways in EF-mediated MEK pathway activation.
Cell permeant Ca2+-chelating agent BAPTA significantly reduced MEK phosphorylation levels in high-frequency EF and no-EF groups (p < 0.05, figure 7b). Interestingly, the phosphorylated MEK levels were lower in the high-frequency EF group when compared with no-EF controls in the presence of BAPTA (p < 0.05, figure 7b), which was similar to the trends in cell responses observed in the presence of MEK inhibitor U0126 (figures 4 and 6), indicating the possible involvement of Ca2+ signalling in regulation of MEK pathway activation by high-frequency EF. These effects were not observed in low-frequency EF groups.
Pre-treatment of endothelial cells with eNOS inhibitor (l-NAME) did not affect the base pMEK levels in no-EF controls (figure 7c). However, inhibition of eNOS by l-NAME abolished the EF-induced increase in pMEK, suggesting that eNOS signalling may play a partial role in EF-mediated MAPK/ERK pathway activation.
3.5. Electric field exposure did not affect endothelial cell apoptosis or proliferation
Caspase-3 staining showed no significant effect of EF on endothelial cell apoptosis (see the electronic supplementary material, figure S4a), with less than 5 per cent apoptotic cells observed in all experimental groups at 12 h as well as 24 h of EF exposure. At 12 h of EF exposure, BrdU staining of endothelial cells indicated a trend of increased cell proliferation with increased frequency, although it was not statistically significant (see the electronic supplementary material, figure S4b). Also there were no significant differences in the number of proliferating cells after 24 h of EF exposure.
3.6. In vivo electric field exposure enhances vascular endothelial growth factor expression in diabetic wounds
For a preliminary in vivo validation of the in vitro results, a mouse db/db model of diabetic wound healing was used [55]. Wound treatment with high-frequency EF (figure 8a) resulted in significantly increased VEGF protein levels in the wound tissue when compared with no-EF-treated control wounds (figure 8b). No detrimental effects of EF exposure on wound healing were observed during EF treatment. Importantly, previous studies demonstrated that increased VEGF expression results in improved healing in diabetic wounds [59], suggesting that high-frequency low-amplitude EF exposure has therapeutic potential. Ongoing studies are focusing on evaluating EF-mediated wound healing and neovascularization in more detail.
Figure 8.

In vivo exposure to high-frequency EF enhances VEGF expression in diabetic wounds: (a) In vivo EF exposure set-up consisted of two antennae, which were connected to the EF source through a flexible co-axial cable. The antennae were approximately 5 mm away from the wounds created on the back of the diabetic mice. EF stimulation was applied for 1 h per day for 7 days. (b) Wound treatment with high-frequency 7.5 GHz EF significantly increased VEGF protein levels in the wound tissue when compared with no-EF treated control wounds (n = 5, p < 0.05).
4. Discussion
The results of this study demonstrate that physiological (low-amplitude) EF can stimulate angiogenic responses in microvascular endothelial cells via a frequency-sensitive VEGFR2-independent activation of the MAPK/ERK pathway. This activation then results in enhanced release of VEGF and may synergistically promote angiogenesis along with the EF stimulation. The in vitro effects are observed in the regime where the EF component is normal to the cell surface and no EF-induced directional cell migration (electrotaxis) is present, suggesting that the pro-angiogenic effect of EF is uncoupled from electrotaxis-related cytoskeletal rearrangements reported in other systems [18].
Importantly, our results provide new information regarding the major roles of both field orientation and frequency in endothelial cell angiogenic responses, which may be critical for choosing optimal field parameters for EF-based pro-angiogenic therapies. In our configuration, high-frequency EF enhanced capillary morphogenesis, MAPK/ERK pathway activation and VEGF release. However, low-frequency (60 Hz) EF did not affect cell responses, and has actually resulted in decreased phosphorylation levels of ERK. These results are in contrast to previous studies, where profound effects of 50 Hz electromagnetic field on angiogenic processes [60] and MAP kinase activation in HL-60 cells [34] were observed, which may be due to the differences in field orientation used in the present study (field normal to the cell surface) and previous studies (field is parallel to the cell surface), field component (electric in our study and magnetic in Monache et al. [60]), or cell type. Previous studies also reported that DC EF of physiological amplitude oriented parallel to the cell plane promoted cell responses such as reorientation, migration, actin assembly and VEGF release in human umbilical cord (HUVEC) and bovine aortic endothelial cells [10,18]. A study by Monache et al. [60] demonstrated that low-frequency (50 Hz) magnetic fields normal to the cell plane enhanced HUVEC angiogenic response in vitro through VEGF- (and ERK)-dependent signal transduction pathways. Another low-frequency EF modality with EF parallel to cell surface (asymmetric 4.5 ms pulses repeated at 15 Hz) stimulated angiogenic response in HUVECs via fibroblast growth factor-2 (FGF-2), but not through the VEGF expression [16,17], and accelerated wound healing under diabetic and normal conditions in vivo by up-regulation of FGF-2-mediated angiogenesis [14]. Interestingly that in vivo, where direction of the field is difficult to control precisely, both 50 Hz electrical stimulation as well as pulse EF (0.3 msec square-wave pulses) significantly enhanced in vivo angiogenesis in both ischaemic and non-ischaemic rat limbs, which was mediated through the increased expression of VEGF [5,12]. These results are consistent with the findings of this study, where high-frequency EF stimulation resulted in increased VEGF expression both in vitro and in the wound tissue in vivo. Overall, our results, together with the previous studies by other groups, suggest that endothelial cells may respond differently to field type (magnetic and/or electric), frequency and orientation. More studies are needed to dissect the mechanisms for such selectivity, which are currently not understood.
In this study, we have focused on the effects of physiological EF on MAPK signalling cascades (figure 9a). Of this family, the ERK pathway is the major regulator of angiogenic responses in endothelial cells [25,29,56], and is activated in response to growth factors (e.g. VEGF), and/or cytokines, radiation and oxidative stress [56,61]. This pathway can also cross-talk with Ca2+ and eNOS [62–64] and PI3K pathways [65,66]. Two other members of the MAPK family—JNK and p38 pathways—are involved in both angiogenic and stress-activated apoptotic signalling, and can also be activated by radiation and oxidative stress [24,26–28,30]. Our results show that high-frequency EF enhances capillary morphogenesis and VEGF release via the mechanism that probably does not require VEGF binding to VEGFR2 receptor, but involves signalling via the ERK pathway and, specifically, MEK protein. MEK is a central regulatory component in the MAPK/ERK pathway [61] and shows high specificity towards upstream cRaf and downstream ERK protein [67]. In the present study, EF does not alter the total MEK levels, which is consistent with previous reports of no effect of applied fields on ERK expression [35]. However, high-frequency EF results in increased MEK–cRaf complex formation, increased MEK phosphorylation levels (overall and within this complex), decreased levels of free MEK and enhanced ERK phosphorylation in endothelial cells, while these responses are not elicited by low-frequency EF.
Figure 9.

(a) Angiogenic signalling pathways and (b) proposed mechanism for high-frequency EF regulation of angiogenic response. Our results demonstrate that high-frequency EF enhances capillary morphogenesis and VEGF release via the MEK–cRaf step in the ERK pathway, with a possible involvement of Ca2+—and, to a lesser extent, eNOS-mediated MEK activation. The observed pro-angiogenic effects of EF are frequency-sensitive and are independent of VEGF binding to VEGFR2 receptor and PI3K signalling. A potential mechanism for these effects is that high-frequency EF may directly regulate the interaction of MEK protein with its binding partner cRaf (b), consistent with the possibility of gigahertz EF penetrating the cell membrane and affecting intracellular signalling. Thus, 7.5 GHz EF may enhance interaction between MEK and its binding partner (c-Raf or U0126), resulting in an increased complex formation, increased phosphorylation of MEK protein and activation of downstream MAPK/ERK pathway, ultimately leading to stimulation of angiogenic responses in the absence of U0126, or inhibition of angiogenic responses when U0126 is present.
The difference in cell responses to high (7.5 GHz) and low (60 Hz) frequencies observed in this study may also be related to the different mechanisms of EF interactions with the cells. It has been suggested that at low frequencies (below 100 MHz), the cell (including cell membrane, cytoplasm and nucleus) can be considered a conductive medium with high membrane impedance, which results in shielding the inside of the cell from the external field. On the other hand, at high frequencies (in the gigahertz range), the membrane impedance becomes negligible, which leads to the flow of current through intracellular space [38,68,69]. In this scenario, 7.5 GHz EF may be able to directly access intracellular space and regulate intracellular processes, such as protein–protein interactions, while 60 Hz EF would not have this capability. Indeed, our results in the presence of the highly specific MEK inhibitor U0126 are consistent with this model. Mechanistically, several studies have shown that MEK phosphorylation and MAPK/ERK pathway activation require MEK binding with upstream cRaf at serine 218 and serine 222 motifs [56,61]. On the one hand, cell exposure to 7.5 GHz EF in our experiments enhanced the formation of MEK–cRaf complexes, decreased free MEK levels and increased MEK phosphorylation levels when compared with 60 Hz or no-EF groups (figure 6), ultimately resulting in stimulation of angiogenic response by endothelial cells. On the other hand, in the presence of U0126, cell exposure to 7.5 GHz EF resulted in significantly decreased formation of MEK-cRaf complex levels and increased free MEK levels when compared with 60 Hz or no-EF groups, effectively abolishing downstream angiogenic response under these conditions. These results can be explained using the model in figure 9b, where 7.5 GHz EF enhances interaction between MEK and its binding partner (c-Raf or U0126), resulting in stimulation of capillary morphogenesis and VEGF release in the absence of U0126 or inhibition of capillary morphogenesis and VEGF release when U0126 is present.
Further studies of the mechanism for EF-mediated MEK phosphorylation show that both Ca2+ chelator and MEK inhibitor lead to significant decreases in MEK phosphorylation and angiogenic responses to high-frequency EF. Reduced MAPK/ERK levels in BAPTA treated samples have been reported previously [53]. It has also been shown that Ca2+ activates PKC [70], which in turn directly phosphorylates MEK [71]. Therefore, it is possible that EF also enhances MEK phosphorylation through Ca2+–PKC–MEK crosstalk, along with activating cRaf–MEK–ERK. These findings suggest a major role for cRaf/MEK and Ca2+ pathways in EF-mediated stimulation of angiogenic responses. These results are also consistent with recent studies in non-endothelial cells which have shown that high-frequency EF can affect intracellular processes, including activation of the ERK1/2 pathway in Rat1 and HeLa cells by a 800–950 MHz electromagnetic field [35], as well as Ca2+ redistribution in Jurkat cells following stimulation with a nanosecond high-amplitude pulse EF [68,69].
In contrast to cRaf/MEK and Ca2+ results, our data show that while inhibition of eNOS with l-NAME does not alter MEK phosphorylation in the absence of EF, it decreases pMEK levels that have been increased by EF to control values. These results are consistent with previous studies that l-NAME does not affect VEGF-induced ERK activity [51,54]. However, the mechanisms for the observed response to eNOS inhibition in the presence of EF are not clear and will be the subject of our future work.
Our results suggest that PI3K pathway does not play a major role in EF-mediated MEK activation in endothelial cells. PI3K in one of the upstream mediators of MEK, and PI3K/Akt signalling can regulate MEK phosphorylation in various cell types. The interactions between the Raf/MEK/ERK and PI3K/Akt pathways occur when Akt regulates Raf activity, which results in the downstream activation of the MEK-ERK pathway [65]. As expected [57], addition of a specific PI3K inhibitor LY294002 results in an overall decrease in pMEK levels (figure 7b); however, the EF-mediated increase in pMEK in the high-frequency group relative to the no EF controls is still retained.
In conclusion, this study provides evidence for a novel mechanism of EF-mediated regulation of endothelial cell angiogenic responses via frequency-sensitive VEGFR2-independent activation of the MAPK/ERK signalling pathway. In vivo, this mechanism translates into VEGF accumulation in the wound, which may result in increased wound vascularization and improved healing [59]. Therefore, these findings may have important implications with regard to the therapeutic use of EF to stimulate vascular tissue regeneration and repair, where the informed choice of the therapeutic field parameters for angiogenic activation of endothelial cells in the chronic wounds is essential. Currently, the FDA-approved use of EF-based devices in the USA is mostly limited to the healing of bone fractures and treatment of pain and oedema and preventing muscle atrophy [6,72]. Our results in combination with other studies provide valuable information regarding how EF of various modalities can affect different steps in pro-angiogenic signal transduction pathways, which expands our understanding of the biophysical interactions between the cell and the surrounding environment. Therefore, the findings of this study may contribute to the technological advancement and the development of new treatment strategies for chronic wound healing and ischaemic vascular disease without introducing systemic effects [8,73].
Acknowledgements
All procedures were approved by the Institutional Animal Care and Use Committee.
The authors acknowledge John Markus for his assistance in building the EF set-up and Zhuting Sun for assistance with numeric EF calculations. This project was supported by NIH/NDDK (1R21DK078814-01A1) and American Heart Association Beginning grant in aid (BGIA- 533 0765425B) to D.A.N., N.S.F. (DMR-1206784) and (DMR 0804199) to A.B.K., Nanomedicine Fellowship (University of Cincinnati Nanomedicine Group) to A.Q.S. and start-up funds from University of Cincinnati Biomedical Engineering (D.A.N.) and Physics (A.B.K.) departments.
References
- 1.Barker A. T., Jaffe L. F., Vanable J. W., Jr 1982. The glabrous epidermis of cavies contains a powerful battery. Am. J. Physiol. 242, R358–R366 [DOI] [PubMed] [Google Scholar]
- 2.Funk R. H. W., Monsees T. K. 2006. Effects of electromagnetic fields on cells: physiological and therapeutical approaches and molecular mechanisms of interaction. Cells Tissues Organs 182, 59–78 10.1159/000093061 (doi:10.1159/000093061) [DOI] [PubMed] [Google Scholar]
- 3.Ieran M., Zaffuto S., Bagnacani M., Annovi M., Moratti A., Cadossi R. 1990. Effect of low frequency pulsing electromagnetic fields on skin ulcers of venous origin in humans: a double-blind study. J. Orthop. Res. 8, 276–282 10.1002/jor.1100080217 (doi:10.1002/jor.1100080217) [DOI] [PubMed] [Google Scholar]
- 4.Salzberg C. A., Cooper-Vastola S. A., Perez F., Viehbeck M. G., Byrne D. W. 1995. The effects of non-thermal pulsed electromagnetic energy on wound healing of pressure ulcers in spinal cord-injured patients: a randomized, double-blind study. Ostomy Wound Manage. 41, 42–44 [PubMed] [Google Scholar]
- 5.Kanno S., Nobuyuki O., Mayumi A., Saito S., Hori K., Handa Y., Tabayashi K., Sato Y. 1999. Establishment of a simple and practical procedure applicable to therapeutic angiogenesis. Circulation 99, 2682–2687 10.1161/01.CIR.99.20.2682 (doi:10.1161/01.CIR.99.20.2682) [DOI] [PubMed] [Google Scholar]
- 6.Akai M., Hayashi K. 2002. Effect of electrical stimulation on musculoskeletal systems; a meta-analysis of controlled clinical trials. Bioelectromagnetics 23, 132–143 10.1002/bem.106 (doi:10.1002/bem.106) [DOI] [PubMed] [Google Scholar]
- 7.Larsen J. A., Overstreet J. 2008. Pulsed radio frequency energy in the treatment of complex diabetic foot wounds: two cases. J. Wound Ostomy Continence Nurs. 35, 523–527 10.1097/01.WON.0000335966.98607.1c (doi:10.1097/01.WON.0000335966.98607.1c) [DOI] [PubMed] [Google Scholar]
- 8.Ojingwa J. C., Isseroff R. R. 2003. Electrical stimulation of wound healing. J. Invest. Dermatol. 121, 1–12 10.1046/j.1523-1747.2003.12454.x (doi:10.1046/j.1523-1747.2003.12454.x) [DOI] [PubMed] [Google Scholar]
- 9.Markov M. S. 2007. Pulsed electromagnetic field therapy history, state of the art and future. Environmentalist 27, 465–475 10.1007/s10669-007-9128-2 (doi:10.1007/s10669-007-9128-2) [DOI] [Google Scholar]
- 10.Zhao M., Bai H., Wang E., Forrester J. V., McCaig C. D. 2004. Electrical stimulation directly induces pre-angiogenic responses in vascular endothelial cells by signaling through VEGF receptors. J. Cell Sci. 117, 397–405 10.1242/jcs.00868 (doi:10.1242/jcs.00868) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrara N., Kerbel R. S. 2005. Angiogenesis as a therapeutic target. Nature 438, 967–974 10.1038/nature04483 (doi:10.1038/nature04483) [DOI] [PubMed] [Google Scholar]
- 12.Linderman J. R., Kloehn M. R., Greene A. S. 2000. Development of an implantable muscle stimulator: measurement of stimulated angiogenesis and poststimulus vessel regression. Microcirculation 7, 119–128 10.1038/sj.mn.7300100 (doi:10.1038/sj.mn.7300100) [DOI] [PubMed] [Google Scholar]
- 13.Roland D., Ferder M., Kothuru R., Faierman T., Strauch B. 2000. Effects of pulsed magnetic energy on a microsurgically transferred vessel. Plast. Reconstr. Surg. 105, 1371–1374 10.1097/00006534-200004040-00016 (doi:10.1097/00006534-200004040-00016) [DOI] [PubMed] [Google Scholar]
- 14.Callaghan M. J., Chang E. I., Seiser N., Aarabi S., Ghali S., Kinnucan E. R., Simon E. R., Gurtner G. C. 2008. Pulsed electromagnetic fields accelerate normal and diabetic wound healing by increasing endogenous FGF-2 release. Plast. Reconstr. Surg. 121, 130–141 10.1097/01.prs.0000293761.27219.84 (doi:10.1097/01.prs.0000293761.27219.84) [DOI] [PubMed] [Google Scholar]
- 15.Bai H., Forrester J. V., Zhao M. 2011. DC electric stimulation upregulates angiogenic factors in endothelial cells through activation of VEGF receptors. Cytokine 55, 110–115 10.1016/j.cyto.2011.03.003 (doi:10.1016/j.cyto.2011.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tepper O. M., et al. 2004. Electromagnetic fields increase in vitro and in vivo angiogenesis through endothelial release of FGF-2. FASEB J. 18, 1231–1233 10.1096/fj.03-0847fje (doi:10.1096/fj.03-0847fje) [DOI] [PubMed] [Google Scholar]
- 17.Hopper R. A., Verhalen J. P., Tepper O. T., Mehrara B. J., Detch R., Chang E. I., Baharestani S., Simon B. J., Gurtner G. C. 2009. Osteoblasts stimulated with pulsed electromagnetic fields increase HUVEC proliferation via a VEGF-A independent mechanism. Bioelectromagnetics 30, 189–97 10.1002/bem.20459 (doi:10.1002/bem.20459) [DOI] [PubMed] [Google Scholar]
- 18.Li X., Kolega J. 2002. Effects of direct current electric fields on cell migration and actin filament distribution in bovine vascular endothelial cells. J. Vasc. Res. 39, 391–404 10.1159/000064517 (doi:10.1159/000064517) [DOI] [PubMed] [Google Scholar]
- 19.Finkelstein E., Chang W., Chao P. H., Gruber D., Minden A., Hung C. T., Finkelstein E. 2004. Roles of microtubules, cell polarity and adhesion in electric-field-mediated motility of 3T3 fibroblasts. J. Cell Sci. 117, 1533–1545 10.1242/jcs.00986 (doi:10.1242/jcs.00986) [DOI] [PubMed] [Google Scholar]
- 20.Rawe I. M., Vlahovic T. C. 2011. The use of a portable, wearable form of pulsed radio frequency electromagnetic energy device for the healing of recalcitrant ulcers: a case report. Int. Wound J. 9, 253–258 10.1111/j.1742-481X.2011.00853.x (doi:10.1111/j.1742-481X.2011.00853.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lappin M. S., Lawrie L. F., Richards T. L., Kramer E. D. 2003. Effects of a pulsed electromagnetic therapy on multiple sclerosis fatigue and quality of life: a double-blind, placebo controlled trial. Altern. Ther. Health Med. 9, 38. [PubMed] [Google Scholar]
- 22.Guo L., Kubat N. J., Isenberg R. A. 2011. Pulsed radio frequency energy (PRFE) use in human medical applications. Electromagn. Biol. Med. 30, 21–45 10.3109/15368378.2011.566775 (doi:10.3109/15368378.2011.566775) [DOI] [PubMed] [Google Scholar]
- 23.Seger R., Krebs E. G. 1995. The MAPK signaling cascade. FASEB J. 9, 726–735 [PubMed] [Google Scholar]
- 24.Rousseau S., Houle F., Landry J., Huot J. 1997. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15, 2169–2177 10.1038/sj.onc.1201380 (doi:10.1038/sj.onc.1201380) [DOI] [PubMed] [Google Scholar]
- 25.Gupta K., Kshirsagar S., Li W., Gui L., Ramakrishnan S., Gupta P., Gupta K. 1999. VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp. Cell Res. 247, 495–504 10.1006/excr.1998.4359 (doi:10.1006/excr.1998.4359) [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto T., Yokote K., Tamura K., Takemoto M., Ueno H., Saito Y., Matsumoto T. 1999. Platelet-derived growth factor activates p38 mitogen-activated protein kinase through a Ras-dependent pathway that is important for actin reorganization and cell migration. J. Biol. Chem. 274, 13 954–13 960 10.1074/jbc.274.20.13954 (doi:10.1074/jbc.274.20.13954) [DOI] [PubMed] [Google Scholar]
- 27.Hu Y. L., Li S., Shyy J. Y., Chien S. 1999. Sustained JNK activation induces endothelial apoptosis: studies with colchicine and shear stress. Am. J. Physiol. 277, H1593–H1599 [DOI] [PubMed] [Google Scholar]
- 28.Deschesnes R. G., Huot J., Valerie K., Landry J. 2001. Involvement of p38 in apoptosis-associated membrane blebbing and nuclear condensation. Mol. Biol. Cell. 12, 1569–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mavria G., Vercoulen Y., Yeo M., Paterson H., Karasarides M., Marais R., Bird D., Marshall C. J. 2006. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell. 9, 33–44 10.1016/j.ccr.2005.12.021 (doi:10.1016/j.ccr.2005.12.021) [DOI] [PubMed] [Google Scholar]
- 30.Medhora M., Dhanasekaran A., Pratt P. F., Jr, Cook C. R., Dunn L. K., Gruenloh S. K., Jacobs E. R. 2008. Role of JNK in network formation of human lung microvascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L676–L785 10.1152/ajplung.00496.2007 (doi:10.1152/ajplung.00496.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leszczynski D., Joenvaara S., Reivinen J., Kuokka R. 2002. Non-thermal activation of the hsp27/p38MAPK stress pathway by mobile phone radiation in human endothelial cells: molecular mechanism for cancer- and blood–brain barrier-related effects. Differentiation 70, 120–129 10.1046/j.1432-0436.2002.700207.x (doi:10.1046/j.1432-0436.2002.700207.x) [DOI] [PubMed] [Google Scholar]
- 32.Bernardini C., Zannoni A., Turba M. E., Bacci M. L., Forni M., Mesirca P., Remondini D., Castellani G., Bersani F. 2007. Effects of 50 Hz sinusoidal magnetic fields on Hsp27, Hsp70, Hsp90 expression in porcine aortic endothelial cells (PAEC). Bioelectromagnetics 28, 231–237 10.1002/bem.20299 (doi:10.1002/bem.20299) [DOI] [PubMed] [Google Scholar]
- 33.Sauer H., Bekhite M. M., Hescheler J., Wartenberg M. 2005. Redox control of angiogenic factors and CD31-positive vessel-like structures in mouse embryonic stem cells after direct current electrical field stimulation. Exp. Cell Res. 304, 380–390 10.1016/j.yexcr.2004.11.026 (doi:10.1016/j.yexcr.2004.11.026) [DOI] [PubMed] [Google Scholar]
- 34.Nie K., Henderson A. 2003. MAP kinase activation in cells exposed to a 60 Hz electromagnetic field. J. Cell. Biochem. 90, 1197–1206 10.1002/jcb.10704 (doi:10.1002/jcb.10704) [DOI] [PubMed] [Google Scholar]
- 35.Friedman J., Kraus S., Hauptman Y., Schiff Y., Seger R. 2007. Mechanism of short-term ERK activation by electromagnetic fields at mobile phone frequencies. Biochem. J. 405, 559–568 10.1042/BJ20061653 (doi:10.1042/BJ20061653) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Zhao M., et al. 2006. Electrical signals control wound healing through phosphatidylinositol-3-OH kinase-γ and PTEN. Nature 442, 457–460 10.1038/nature04925 (doi:10.1038/nature04925) [DOI] [PubMed] [Google Scholar]
- 37.Cho M. R., Thatte H. S., Lee R. C., Golan D. E. 1996. Reorganization of microfilament structure induced by ac electric fields. FASEB J. 10, 1552–1558 [DOI] [PubMed] [Google Scholar]
- 38.Foster K. R. 2000. Thermal and nonthermal mechanisms of interaction of radio-frequency energy with biological systems. IEEE Trans. Plasma Sci. 28, 15–23 10.1109/27.842819 (doi:10.1109/27.842819) [DOI] [Google Scholar]
- 39.Lim Y. C., Garcia-Cardena G., Allport J. R., Zervoglos M., Connolly A. J., Gimbrone M. A., Jr, Luscinskas F. W. 2003. Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am. J. Pathol. 162, 1591. 10.1016/S0002-9440(10)64293-9 (doi:10.1016/S0002-9440(10)64293-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bahr A., Dorn H., Bolz T. 2006. Dosimetric assessment of an exposure system for simulating GSM and WCDMA mobile phone usage. Bioelectromagnetics 27, 320–327 10.1002/bem.20218 (doi:10.1002/bem.20218) [DOI] [PubMed] [Google Scholar]
- 41.Cho H., Balaji S., Sheikh A. Q., Hurley J. R., Tian Y. F., Collier J. H., Crombleholme T. M., Narmoneva D. A. 2011. Regulation of endothelial cell activation and angiogenesis by injectable peptide nanofibers. Acta Biomater. 8, 154–164 10.1016/j.actbio.2011.08.029 (doi:10.1016/j.actbio.2011.08.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurley J. R., Balaji S., Narmoneva D. A. 2010. Complex temporal regulation of capillary morphogenesis by fibroblasts. Am. J. Physiol. Cell Physiol. 299, C444–C453 10.1152/ajpcell.00572.2009 (doi:10.1152/ajpcell.00572.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narmoneva D. A., Oni O., Sieminski A. L., Zhang S., Gertler J. P., Kamm R. D., Lee R. T. 2005. Self-assembling short oligopeptides and the promotion of angiogenesis. Biomaterials 26, 4837. 10.1016/j.biomaterials.2005.01.005 (doi:10.1016/j.biomaterials.2005.01.005) [DOI] [PubMed] [Google Scholar]
- 44.Autiero M., Luttun A., Tjwa M., Carmeliet P. 2003. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J. Thromb. Haemost. 1, 1356–1370 10.1046/j.1538-7836.2003.00263.x (doi:10.1046/j.1538-7836.2003.00263.x) [DOI] [PubMed] [Google Scholar]
- 45.Shibuya M. 2006. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J. Biochem. Mol. Biol. 39, 469–748 10.5483/BMBRep.2006.39.5.469 (doi:10.5483/BMBRep.2006.39.5.469) [DOI] [PubMed] [Google Scholar]
- 46.Craig T. J., Ciufo L. F., Morgan A. 2004. A protein–protein binding assay using coated microtitre plates: increased throughput, reproducibility and speed compared to bead-based assays. J. Biochem. Biophys. Methods 60, 49–60 10.1016/j.jbbm.2004.04.015 (doi:10.1016/j.jbbm.2004.04.015) [DOI] [PubMed] [Google Scholar]
- 47.Adler V., et al. 2005. Functional interactions of raf and MEK with Jun-N-terminal kinase (JNK) result in a positive feedback loop on the oncogenic ras signaling pathway. Biochemistry 44, 10 784–10 795 10.1021/bi050619j (doi:10.1021/bi050619j) [DOI] [PubMed] [Google Scholar]
- 48.Cheng C., et al. 2009. Trihydrophobin 1 interacts with PAK1 and regulates ERK/MAPK activation and cell migration. J. Biol. Chem. 284, 8786–8796 10.1074/jbc.M806144200 (doi:10.1074/jbc.M806144200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai J., Hong Y., Weng C., Tan C., Imperato-McGinley J., Zhu Y. S. 2011. Androgen stimulates endothelial cell proliferation via an androgen receptor/VEGF/cyclin A-mediated mechanism. Am. J. Physiol. Heart Circ. Physiol. 300, H1210–H1221 10.1152/ajpheart.01210.2010 (doi:10.1152/ajpheart.01210.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang K. S., Ionides E., Oster G., Nuccitelli R., Isseroff R. R. 1999. Epidermal growth factor receptor relocalization and kinase activity are necessary for directional migration of keratinocytes in DC electric fields. J. Cell Sci. 112, 1967–1978 [DOI] [PubMed] [Google Scholar]
- 51.Gliki G., Abu-Ghazaleh R., Jezequel S., Wheeler-Jones C., Zachary I. 2001. Vascular endothelial growth factor-induced prostacyclin production is mediated by a protein kinase C (PKC)-dependent activation of extracellular signal-regulated protein kinases 1 and 2 involving PKC-δ and by mobilization of intracellular Ca2+. Biochem. J. 353, 503–512 10.1042/0264-6021:3530503 (doi:10.1042/0264-6021:3530503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vlahos C. J., Matter W. F., Hui K. Y., Brown R. F. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241–5248 [PubMed] [Google Scholar]
- 53.Momberger T. S., Levick J. R., Mason R. M. 2006. Mechanosensitive synoviocytes: A Ca2+-PKCα-MAP kinase pathway contributes to stretch-induced hyaluronan synthesis in vitro. Matrix Biol. 25, 306–316 10.1016/j.matbio.2006.01.008 (doi:10.1016/j.matbio.2006.01.008) [DOI] [PubMed] [Google Scholar]
- 54.Breslin J. W., Pappas P. J., Cerveira J. J., Hobson Ii R. W., Duran W. N. 2003. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 284, H92–H100 [DOI] [PubMed] [Google Scholar]
- 55.Keswani S. G., Katz A. B., Lim F. Y., Zoltick P., Radu A., Alaee D., Herlyn M., Crombleholme T. M. 2004. Adenoviral mediated gene transfer of PDGF-B enhances wound healing in type I and type II diabetic wounds. Wound Repair Regen. 12, 497–504 10.1111/j.1067-1927.2004.12501.x (doi:10.1111/j.1067-1927.2004.12501.x) [DOI] [PubMed] [Google Scholar]
- 56.Roux P. P., Blenis J. 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68, 320–344 10.1128/MMBR.68.2.320-344.2004 (doi:10.1128/MMBR.68.2.320-344.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bancroft C. C., Chen Z., Yeh J., Sunwoo J. B., Yeh N. T., Jackson S., Jackson C., Van Waes C. 2002. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-κB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int. J. Cancer 99, 538–548 10.1002/ijc.10398 (doi:10.1002/ijc.10398) [DOI] [PubMed] [Google Scholar]
- 58.Fong T. A. T., et al. 1999. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 59, 99–106 [PubMed] [Google Scholar]
- 59.Galiano R. D., Tepper O. M., Pelo C. R., Bhatt K. A., Callaghan M., Bastidas N., Bunting S., Steinmetz H. G., Gurtner G. C. 2004. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 164, 1935–1947 10.1016/S0002-9440(10)63754-6 (doi:10.1016/S0002-9440(10)63754-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Monache D. S., Alessandro R., Iorio R., Gualtieri G., Colonna R. 2008. Extremely low frequency electromagnetic fields (ELF-EMFs) induce in vitro angiogenesis process in human endothelial cells. Bioelectromagnetics 29, 640–648 10.1002/bem.20430 (doi:10.1002/bem.20430) [DOI] [PubMed] [Google Scholar]
- 61.Rubinfeld H., Seger R. 2005. The ERK cascade: a prototype of MAPK signaling. Mol. Biotechnol. 31, 151–174 10.1385/MB:31:2:151 (doi:10.1385/MB:31:2:151) [DOI] [PubMed] [Google Scholar]
- 62.Bernier S. G., Haldar S., Michel T. 2000. Bradykinin-regulated interactions of the mitogen-activated protein kinase pathway with the endothelial nitric-oxide synthase. J. Biol. Chem. 275, 30 707–30 715 10.1074/jbc.M005116200 (doi:10.1074/jbc.M005116200) [DOI] [PubMed] [Google Scholar]
- 63.Chen Y., Medhora M., Falck J. R., Pritchard K. A., Jr, Jacobs E. R. 2006. Mechanisms of activation of eNOS by 20-HETE and VEGF in bovine pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L378–L385 10.1152/ajplung.00424.2005 (doi:10.1152/ajplung.00424.2005) [DOI] [PubMed] [Google Scholar]
- 64.Chuderland D., Marmor G., Shainskaya A., Seger R. 2008. Calcium-mediated interactions regulate the subcellular localization of extracellular signal-regulated kinases. J. Biol. Chem. 283, 11 176–11 188 10.1074/jbc.M709030200 (doi:10.1074/jbc.M709030200) [DOI] [PubMed] [Google Scholar]
- 65.McCubrey J. A., et al. 2006. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 283, 249–279 10.1016/j.advenzreg.2006.01.004 (doi:10.1016/j.advenzreg.2006.01.004) [DOI] [PubMed] [Google Scholar]
- 66.Steelman L. S., Pohnert S. C., Shelton J. G., Franklin R. A., Bertrand F. E., McCubrey J. A. 2004. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 18, 189–218 10.1038/sj.leu.2403241 (doi:10.1038/sj.leu.2403241) [DOI] [PubMed] [Google Scholar]
- 67.Alessi D. R., Saito Y., Campbell D. G., Cohen P., Sithanandam G., Rapp U., Ashworth A., Marhsall C. J., Cowley S. 1994. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74(raf-1). EMBO J. 13, 1610–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vernier P. T., Sun Y., Marcu L., Salemi S., Craft C. M., Gundersen M. A. 2003. Calcium bursts induced by nanosecond electric pulses. Biochem. Biophys. Res. Commun. 310, 286–295 10.1016/j.bbrc.2003.08.140 (doi:10.1016/j.bbrc.2003.08.140) [DOI] [PubMed] [Google Scholar]
- 69.Beebe S. J., Blackmore P. F., White J., Joshi R. P., Schoenbach K. H. 2004. Nanosecond pulsed electric fields modulate cell function through intracellular signal transduction mechanisms. Physiol. Meas. 25, 1077–1093 10.1088/0967-3334/25/4/023 (doi:10.1088/0967-3334/25/4/023) [DOI] [PubMed] [Google Scholar]
- 70.Clapham D. E. 2007. Calcium signaling. Cell 131, 1047–1058 10.1016/j.cell.2007.11.028 (doi:10.1016/j.cell.2007.11.028) [DOI] [PubMed] [Google Scholar]
- 71.Nelson T. J., Sun M. K., Hongpaisan J., Alkon D. L. 2008. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 585, 76–87 10.1016/j.ejphar.2008.01.051 (doi:10.1016/j.ejphar.2008.01.051) [DOI] [PubMed] [Google Scholar]
- 72.Ay S., Evcik D. 2009. The effects of pulsed electromagnetic fields in the treatment of knee osteoarthritis: a randomized, placebo-controlled trial. Rheumatol. Int. 29, 663–666 10.1007/s00296-008-0754-x (doi:10.1007/s00296-008-0754-x) [DOI] [PubMed] [Google Scholar]
- 73.Patterson C., Runge M. S. 1999. Therapeutic angiogenesis the new electrophysiology. Circulation 99, 2614–2616 10.1161/01.CIR.99.20.2614 (doi:10.1161/01.CIR.99.20.2614) [DOI] [PubMed] [Google Scholar]