

Abstract
ChIPseq is a widely used technique for investigating protein-DNA interactions. Read density profiles are generated by using next-sequencing of protein-bound DNA and aligning the short reads to a reference genome. Enriched regions are revealed as peaks, which often differ dramatically in shape, depending on the target protein1. For example, transcription factors often bind in a site- and sequence-specific manner and tend to produce punctate peaks, while histone modifications are more pervasive and are characterized by broad, diffuse islands of enrichment2. Reliably identifying these regions was the focus of our work.
Algorithms for analyzing ChIPseq data have employed various methodologies, from heuristics3-5 to more rigorous statistical models, e.g. Hidden Markov Models (HMMs)6-8. We sought a solution that minimized the necessity for difficult-to-define, ad hoc parameters that often compromise resolution and lessen the intuitive usability of the tool. With respect to HMM-based methods, we aimed to curtail parameter estimation procedures and simple, finite state classifications that are often utilized.
Additionally, conventional ChIPseq data analysis involves categorization of the expected read density profiles as either punctate or diffuse followed by subsequent application of the appropriate tool. We further aimed to replace the need for these two distinct models with a single, more versatile model, which can capably address the entire spectrum of data types.
To meet these objectives, we first constructed a statistical framework that naturally modeled ChIPseq data structures using a cutting edge advance in HMMs9, which utilizes only explicit formulas-an innovation crucial to its performance advantages. More sophisticated then heuristic models, our HMM accommodates infinite hidden states through a Bayesian model. We applied it to identifying reasonable change points in read density, which further define segments of enrichment. Our analysis revealed how our Bayesian Change Point (BCP) algorithm had a reduced computational complexity-evidenced by an abridged run time and memory footprint. The BCP algorithm was successfully applied to both punctate peak and diffuse island identification with robust accuracy and limited user-defined parameters. This illustrated both its versatility and ease of use. Consequently, we believe it can be implemented readily across broad ranges of data types and end users in a manner that is easily compared and contrasted, making it a great tool for ChIPseq data analysis that can aid in collaboration and corroboration between research groups. Here, we demonstrate the application of BCP to existing transcription factor10,11 and epigenetic data12 to illustrate its usefulness.
Keywords: Genetics, Issue 70, Bioinformatics, Genomics, Molecular Biology, Cellular Biology, Immunology, Chromatin immunoprecipitation, ChIP-Seq, histone modifications, segmentation, Bayesian, Hidden Markov Models, epigenetics
Protocol
1. Preparing Input Files for BCP Analysis
Align the short reads produced from sequencing runs (ChIP and input libraries) to the appropriate reference genome using the preferred short read alignment software. The mapped locations should be converted to the 6 column browser extensible data (BED) format13 (UCSC genome browser, http://genome.ucsc.edu/), a tab-delimited line per mapped read indicating the mapped chromosome, start position (0-based), end position (half-open), read name, score (optional), and strand.
2a. Diffuse Read Profiles: Preprocessing ChIP Read Densities for Detection of Enriched Islands in Diffuse Data
Extend the ChIP and input mapped locations to a predetermined fragment length, i.e. the fragment size targeted during enzyme digestion or sonication of the DNA, usually around 200 bp. Fragment counts are then aggregated in adjacent bins. By default, bin size is set to the estimated fragment length of 200 bp.
Any possible change-points in a set of bins with identical read counts will most likely fall at the outer most boundaries. Accordingly, it is improbable that a change point will occur at an internal boundary between two bins with the same read counts. So, group adjacent bins, with identical reads per bin, into a single block, i.e. bedGraph format13.
2b. Punctate Read Profiles: Preprocessing ChIP and Input BED Files for Detection of Peaks in Punctate Data
Aggregate overlapping reads for plus and minus strand ChIP reads separately. The strand specific read densities should form a bimodal profile of plus and minus peaks. Choose plus/minus pairs of the most enriched peaks and use the distance between their summits as an estimate for the library fragment length.
Shift the ChIP and input reads half the fragment length to the center and recalculate the read density of the shifted and merged plus and minus strand reads. This methodology for estimating the fragment length was adopted from Zhang, et al.3. Positions with identical merge counts should be grouped into blocks, similar to step 2a.2.
3. Estimate the Posterior Mean Read Density of Each Block using our BCMIX Approximation
The read density of each block is modeled as a Poisson distribution, Pois(θt), with a mean parameter following a mixture of Gamma distributions, Γ(α,β), and a prior probability of a change point occurring at any block boundary of p. Conditioning Pois(θt) on G(α,β) effectively renders the model an infinite state HMM. Estimate the hyper-parameters, α, β, and p, using maximum posterior likelihood.
Explicitly calculate the Bayes estimates for each block, θt, as E(θt|γZ). Replace the more traditional but time consuming forward and backward filters often used in HMMs, with the more computationally efficiently Bounded Complexity Mixture approximation to estimate posterior means, θc. The resulting posterior means will be "smoothed" into an approximate piecewise constant profile so blocks with identical, θc, should be further blocked together with updated boundary coordinates.
4a. Diffuse Read Profiles: Post-process Posterior Means into Segments of Diffuse Enrichment
Use the number of input reads per each newθc block as the background rate, Pois(λa) and determine enrichment using a simple hypothesis test based on whether the ChIP posterior mean, θc, exceeds some threshold δ. The 90th-quantile is the default d and is appropriate in most cases.
Merge adjacent θc blocks that exceed the enrichment into a single region and report merge coordinates in simple BED format. Alternatively, one can report the θc for each block in bedGraph format to preserve the high-resolution details of the read density estimates.
4b. Punctate Read Profiles: Post-process Posterior Means into Peak Candidates
Define the background rate, Pois(λa), as the average of all read counts (γ2) and identify all blocks which exceed the threshold, d. Since punctate peaks are expected to be more substantially enriched, the default δ is set to the 99th-quantile of Pois(λa).
Set the block with the maximal θc as the candidate peak summit and adjoin flanking blocks that share a similar read density (±1 read count to allow for slight variation). This adjoined region is defined as a candidate binding site.
Calculateλ2 as the average read counts in the ChIP candidate binding site and hypothesis test this versus input background were the null hypothesis, H0, is that λ1≥λ2 and reject H0based on a p-value threshold. Output candidate peaks in BED format.
Representative Results
BCP excels at identifying regions of broad enrichment in histone modification data. As a point of reference, we previously compared our results to those of SICER3, an existing tool which has demonstrated strong performance. To best illustrate BCP's advantages, we examined a histone modification that had been well studied to establish a foundation for assessing success rates. With this in mind, we then analyzed H3K36me3, since it has been shown to associate strongly with actively transcribed gene bodies (Figure 1). In contrast, H3K36me3 had also been shown to be mutual exclusive to H3K27me3 repressive marks. We further leveraged these known relationships to illustrate the performance advantages of BCP in the accuracy of island calls by determining the fraction of overlap with known associations and disassociations, in effect correlation and anti-correlation. Here, we further substantiate the advantages of BCP using additional examples of high performance.
Our preceding work demonstrated a tendency for much larger island size in BCP, 23.9 to 25.8 kb, than SICER, 2.7 to 10.7 kb; larger islands being more in line with the conventional expectation of broad diffuse islands of H3K36me3 enrichment(PLoS Comp Bio, submitted). Of course, larger islands do not alone indicate accuracy. So, we determined how much overlap these regions had with known genes and contrasted this with the degree of overlap with intergenic space, an indication of false positive rate (FPR). Gene coverage in BCP ranged from 0.492 to 0.497 compared to 0.276 to 0.437 in SICER without severely impacting the FPR; intergenic overlap range from 0.89 to 0.90 and from 0.85 to 0.98 in BCP and SICER, respectively. Here, we present an additional representative region displaying the close relationship between the boundaries of enrichment and gene bodies-clearly distinguishing active and repressed transcription (Figure 1). This further supports our claim that BCP maintains the high overlap of active genes by H3K36me3 islands with boundaries closely aligned to gene bodies without increasing the degree of false positive overlap with intergenic space, genes with repressed transcription, or the H3K27me3 repressive mark.
While assessing the reproducibility of BCP-island calls in two replicate data sets, we noticed BCP did not suffer from a heavy dependence on read coverage depth in the competing algorithm, SICER. We provide additional evidence of BCP's robustness and reproducibility by examining additional distinct regions demonstrating consistent island boundaries despite the reduced coverage depth (simulated by sampling reads from the full data set) (Figure 2).
To fully demonstrate the versatility of BCP, we obtained a broad spectrum of histone modification data, including the punctate marks H3K27ac, H3K9ac, and H3K4me3, and the diffuse mark, H3K9me3, in addition to H3K27me3 and H3K36me3. We analyzed these data sets using the default parameter settings for both BCP and SICER (Figure 3). These marks represent a broad range of read density profiles and allow us to focus on a region that illustrates many of the features commonly associated with them. At the center lies H3K36me3 enrichment at the PXDN gene marking active transcription. Falling expectedly at the transcription start site are the additional punctate, active marks, H3K27ac, H3K9ac, and H3K4me3. Just downstream of PXDN is repressed intergenic space marked by H3K27me3 enrichment. On the opposite flank lies an H3K27me3 repressed gene. Moving one more step out are silenced chromatin, as indicated by the presence of H3K9me3 enrichment which appears to indicate silencing of SNTG2 and MYT1L, perhaps in a less transient sense then H3K27me3 repression. This region encompasses the majority of phenomena encountered in ChIPseq of histone modifications and illustrates how the dynamic nature of BCP can identify both punctate acetylation and H3K4me3 marks while at the same time distinguishing large contiguous islands of H3K27me3 and H3K9me3 repression and H3K36me3 active transcription. To reiterate, BCP can do such all of these analyses simply at default settings and, as demonstrated, still produce quality results, regardless of data type. The algorithm is also fast and memory efficient and, thus, provides a practically compelling usefulness.
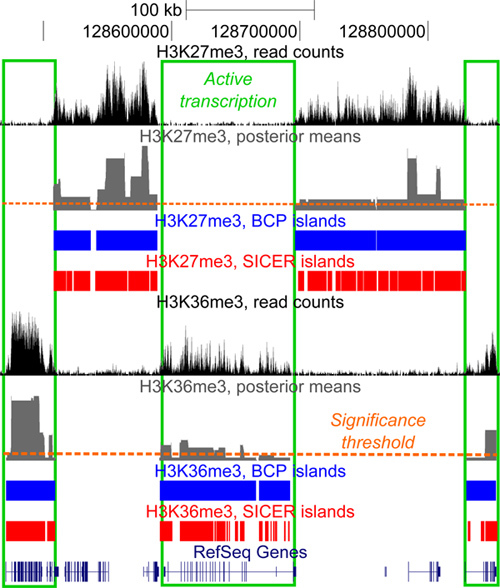 Figure 1. Diffuse read density profiles of histone modifications. H3K27me3 (top) and H3K36me3 (bottom) exemplify the broad, diffuse enrichment islands strongly associated with gene bodies (green boxes). H3K27me3 correlates with repressed genes and intergenic space and anticorrelates with actively transcribed gene bodies. The opposite is true for H3K36me3. Data is visualized in the UCSC genome browser (http://genome.ucsc.edu).
Figure 1. Diffuse read density profiles of histone modifications. H3K27me3 (top) and H3K36me3 (bottom) exemplify the broad, diffuse enrichment islands strongly associated with gene bodies (green boxes). H3K27me3 correlates with repressed genes and intergenic space and anticorrelates with actively transcribed gene bodies. The opposite is true for H3K36me3. Data is visualized in the UCSC genome browser (http://genome.ucsc.edu).
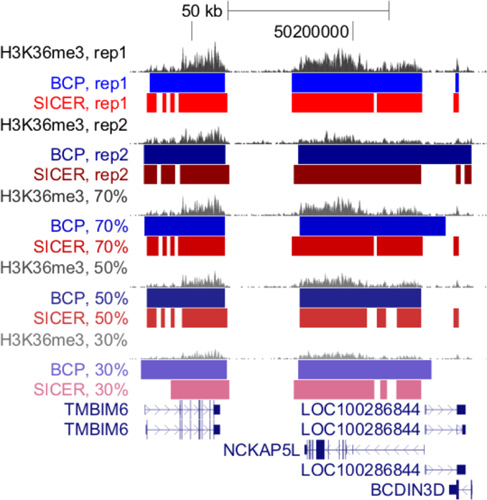 Figure 2. BCP is robust and reproducible. Island calls for H3K36me3 in two replicates and at sampling depths of 30, 50 and 70% of the full replicate 1 dataset were analyzed with BCP. The second replicate, with a substantially lower read coverage, produced similar island calls and the degree of overlap was highly retained regardless of sampling percentage. Furthermore, the islands remained accuracy as seen in the close alignment of boundaries with RefSeq gene body annotations.
Figure 2. BCP is robust and reproducible. Island calls for H3K36me3 in two replicates and at sampling depths of 30, 50 and 70% of the full replicate 1 dataset were analyzed with BCP. The second replicate, with a substantially lower read coverage, produced similar island calls and the degree of overlap was highly retained regardless of sampling percentage. Furthermore, the islands remained accuracy as seen in the close alignment of boundaries with RefSeq gene body annotations.
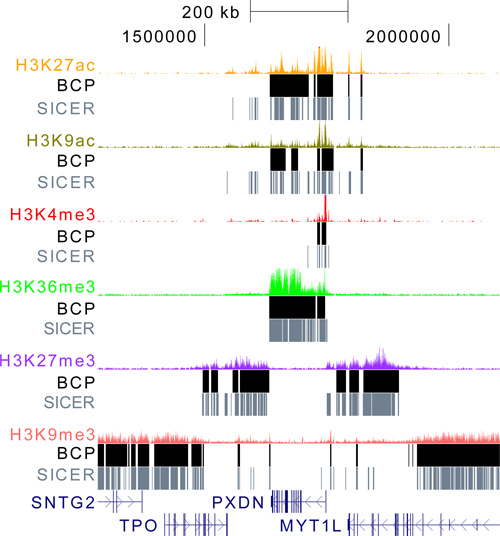 Figure 3. BCP is a versatile algorithm that can be applied to all histone modifications data types. BCP and SICER were used to analyze the gamut of data types, from punctate marks like H3K27ac, H3K9ac, and H3K4me3, to diffuse marks like H3K36me3, H3K27me3, and H3K9me3. Using the default parameters for both algorithms, BCP islands capture the enriched density regardless of their breadth while SICER often fragments regions into many sub-islands. Even in the highly broad and diffuse case of H3K9me3, BCP has reasonable performance.
Figure 3. BCP is a versatile algorithm that can be applied to all histone modifications data types. BCP and SICER were used to analyze the gamut of data types, from punctate marks like H3K27ac, H3K9ac, and H3K4me3, to diffuse marks like H3K36me3, H3K27me3, and H3K9me3. Using the default parameters for both algorithms, BCP islands capture the enriched density regardless of their breadth while SICER often fragments regions into many sub-islands. Even in the highly broad and diffuse case of H3K9me3, BCP has reasonable performance.
Discussion
We set out to develop a model for analyzing ChIPseq data that could identify both punctate and diffuse data structures equally well. Until now, regions of enrichment, particularly diffuse regions, which reflect the presupposed expectation of large island size, have been difficult to identify. To address these problems, we utilized the most recent advances in HMM technology, which possess many advantages over existing heuristic models and less innovative HMMs.
Our model makes use of a Bayesian framework with explicit formulas. This is a crucial distinction from other HMMs, in that it enables us to calculate posterior means, the expected read density of each segment, with simple calculations, rather than relying on time-consuming and computationally costly simulations such as Markov chain Monte Carlo methods. Consequently, our computation times and memory requirements are dramatically reduced. Using high performance compute clusters with dual core, 2.0 Ghz nodes with 2 GB of 64-bit memory to analyze ~23 million H3K27me3 reads or ~21 million H3K36me3 reads, BCP took less than an hour for whole genome analysis compared to several hours to days required for other methods. These timesavings can be achieved with only the modest 2 GB of memory.
Additionally, our model conditions the various means of each segment, i.e. Pois(θ), on a continuous Gamma distribution. Essentially, this allows for infinite possible states for each segment. BCP can provide more than simple binary classifications of enriched versus background and preserves the read density magnitudes for every segment via the output posterior means.
We also make use of the BCMIX algorithm for computational efficiency. This enables a near exhaustive search for change-points between enrichment and background of all possible genomic positions. This provides a heightened resolution not confined by arbitrary window definitions, with little impact on run time or memory demands.
This is all accomplished without perturbing accuracy, both in theory, since the model is statistically rigorous and its results converge to the Bayesian estimator, as well in practice, as we have demonstrated here. The gene coverage of our H3K36me3 results suggest the island calls are highly accurate without encroaching into known mutually excluded intergenic space or H3K27me3 enrichment. The results are remarkably reproducible and robust and showed little dependence on coverage depth, calling similar islands with high gene coverage and low FPR despite sampling depths as low as 30%. BCP was used broadly, without any adjustment to default parameters, to analyze a wide array of histone modification and transcription factor ChIPseq data and performed well in all cases. We hope that due to its high accuracy, robustness, and reproducibility, BCP will serve as an effective tool for data analysis, collaboration, and corroboration in the future.
Disclosures
No conflicts of interest declared.
Acknowledgments
STARR foundation award (MQZ), NIH grant ES017166 (MQZ), NSF grant DMS0906593 (HX).
References
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZS, et al. HPeak: an HMM-based algorithm for defining read-enriched regions in ChIP-Seq data. BMC Bioinformatics. 2010;11:369. doi: 10.1186/1471-2105-11-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Smith AD. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics. 2011;27:870–871. doi: 10.1093/bioinformatics/btr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyrou C, Stark R, Lynch AG, Tavaré S. BayesPeak: Bayesian analysis of ChIP-seq data. BMC Bioinformatics. 2009;10:299. doi: 10.1186/1471-2105-10-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai T, Xing H. A simple Bayesian approach to multiple change-points. Statistica Sinica. 2011.
- Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Stitzel ML, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12:443–455. doi: 10.1016/j.cmet.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:493–496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portales-Casamar E, et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]