

Abstract
Thrombin activates platelets through protease activated receptors (PARs). Mouse platelets express PAR3 and PAR4. PAR3 does not signal in platelets. However, PAR4 is a relatively poor thrombin substrate and requires PAR3 as a cofactor at low thrombin concentrations. In this study we show that PAR3 also regulates PAR4 signaling. In response to thrombin (30–100 nM) or PAR4 activating peptide (AYPGKF), platelets from PAR3−/− mice had increased Gq signaling compared to wild type mice as demonstrated by a 1.6-fold increase in the maximum intracellular calcium (Ca2+) mobilization, an increase in phosphorylation level of protein kinase C (PKC) substrates, and a 2-fold increase of Ca2+ release from intracellular stores. Moreover, platelets from heterozygous mice (PAR3+/−) had an intermediate increase in maximum Ca2+ mobilization. Treatment of PAR3−/− mice platelets with P2Y12 antagonist (2MeSAMP) did not affect Ca2+ mobilization from PAR4 in response to thrombin or AYPGKF. The activation of RhoA-GTP downstream G12/13 signaling in response to thrombin was not significantly different between wild type and PAR3−/− mice. Since PAR3 influenced PAR4 signaling independent of agonist, we examined the direct interaction between PAR3 and PAR4 with bioluminescence resonance energy transfer (BRET). PAR3 and PAR4 form constitutive homodimers and heterodimers. In summary, our results demonstrate that in addition to enhancing PAR4 activation at low thrombin concentrations, PAR3 negatively regulates PAR4-mediated maximum Ca2+ mobilization and PKC activation in mouse platelets by physical interaction.
Introduction
Thrombin signaling in platelets is mediated by protease activated receptors (PARs). PARs are G-protein-coupled receptors (GPCR) that are activated via proteolytic cleavage of the extracellular N-terminus to initiate a variety of signaling cascades through activation of heterotrimeric G proteins [1], [2]. The expression of PARs in platelets is species specific. Human platelets express PAR1 and PAR4 and cleavage of each receptor initiates signaling cascades [3]. Mouse platelets express PAR3 and PAR4, but PAR3 does not signal making PAR4 the signaling receptor [4]–[6].
PAR1 and PAR4 in human platelets are coupled to Gq and G12/13 [7], [8]. The presence of a direct interaction between PARs and Gi is controversial. It has been shown that PAR1 is directly coupled to Gi in human platelets and in COS7 cells transfected with PAR1 [9], [10]. However, other studies have shown that PAR1 and PAR4 do not couple directly to Gi, rather activation of the Gi pathway is mediated by secondary release of ADP, which acts on the Gi-coupled ADP receptor, P2Y12 [8], [11], [12].
A common feature of PAR4 across species is that, on its own, PAR4 is not an efficient thrombin substrate [13]–[15]. As a result, PAR1 in human platelets or PAR3 in mouse platelets serves as a cofactor for PAR4 activation at low thrombin concentrations (<10 nM). However, at high concentrations of thrombin (≥30 nM), PAR4 is sufficient to induce platelet activation [6]. Two independent studies show that PAR3 can affect PAR4 signaling, Nakanishi-Matsui et al, reported that the amount of accumulated inositol phosphate (IP) in response to thrombin (10–100 nM) was 1.7-fold increased in COS7 cells expressing mouse PAR4 alone compared to COS7 cells expressing mouse PAR4 and PAR3 [6]. In addition, Mao et al. showed an increase in intracellular Ca2+ mobilization and platelet aggregation in response to plasmin, in PAR3 knockout (PAR3−/−) mouse platelets compared to wild type [16]. These studies show that PAR3 can influence PAR4 signaling in addition to enhancing PAR4 activation. There are also examples of PAR3 regulating signaling from other PAR family members in endothelial cells and podocytes [17], [18].
In the present study we aimed to determine if the activation of PAR4 with thrombin concentrations that occur at the site of the growing thrombus [19] is affected by the presence of PAR3 in mouse platelets. We report here that PAR3 negatively regulates PAR4-mediated Gq signaling by down regulation of Ca2+ mobilization and PKC activation without affecting the G12/13 pathway as measured by RhoA activation. The negative regulation of PAR3 on PAR4 signaling was independent of the PAR4 agonist. Therefore, we examined the physical interaction between PAR3 and PAR4 with bioluminescence resonance energy transfer (BRET). We also show for the first time that PAR3 forms a constitutive heterodimer with PAR4, and this interaction may affect PAR4 signaling when PAR3 is absent. The results from this study demonstrate that PAR4 signaling can be modulated by other PAR subtypes at thrombin concentrations that are found in vivo at the site of the thrombus. This may have important implications for PAR4 signaling in human platelets where it is co-expressed with PAR1. More generally, the physical interaction between platelet GPCRs may provide unique signaling and may have broad implications for the design of antiplatelet agents.
Materials and Methods
Reagents and Antibodies
Human α-thrombin (specific activity of 5380 NIH units/mg) was purchased from Haematological Technologies (Essex Junction, VT). PAR4 activating peptide (AYPGKF-NH2) was synthesized at PolyPeptide Laboratories (San Diego, CA). Convulxin was purchased from Enzo Life Sciences Inc. (Farmingdale, NY). ADP was purchased from Chrono-log Corporation (Havertown, PA). Fura-2AM and all cell culture reagents were purchased from Invitrogen. Prostaglandin I2 was purchased from Calbiochem. Heparin, thapsigargin, and 2-Methylthioadenosine 5′-monophosphate triethylammonium salt hydrate (2MeSAMP) were purchased from Sigma Chemical Co. The anti-phospho-(Ser) PKC substrates, anti-PKC, anti-phospho-Akt (Ser473) antibodies were purchased from Cell Signaling Technology Inc. (Danvers, MA). The anti-α-actinin antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The anti-PAR4-FITC antibody was purchased from Alamone Labs Ltd. (Jerusalem, Israel). The anti-HA tag Alexa Fluor 647 (6E2) antibody was purchased from Cell Signaling Technology Inc. (Danvers, MA). The anti-V5 tag Alexa Fluor 647 antibody was purchased from AbD Serotec. (Raleigh, NC).
Animals
PAR3 knockout (PAR3−/−) and PAR3 heterozygous (PAR3+/−) mice have been described and were obtained from Mutant Mouse Regional Resource Center (MMRRC, Chapel Hill, NC) [5]. All animal studies were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University School of Medicine.
Platelet preparation
Mice were anesthetized with intraperitoneal injection of pentobarbital (62 mg/kg). Blood was collected from mice by heparinized capillary puncture of the retro-orbital venous sinus and immediately combined with (1/5) volume of acid citrate dextrose (ACD) as an anticoagulant. The whole blood was centrifuged at 2300× g for 20 sec at room temperature (RT) to isolate platelet-rich plasma (PRP). The platelets were pelleted and washed once at 2200× g for 3 min at RT in HEPES-Tyrode buffer pH 7.4 (10 mM HEPES, 12 mM NaHCO3, 130 mM NaCl, 5 mM KCl, 0.4 mM Na2HPO4, 1 mM MgCl2, 5 mM glucose, 0.33% w/v human serum albumin) containing 0.5 µM prostaglandin I2 (PGI2) and 10 U/mL Heparin. Washed platelets were counted on a Hemavet 950FS (Drew Scientific Inc, Waterbury, CT, USA) and the final platelet count adjusted with HEPES-Tyrode's buffer.
Measurement of the concentration of free intracellular Ca2+ ([Ca2+]i)
Washed mouse platelets adjusted to a final concentration of 2×108 platelets/mL were loaded with 10 µM Fura-2 for 45 minutes at room temperature. Platelets were washed once and resuspended to their original concentration in HEPES-Tyrode buffer (pH 7.4) containing 2 mM CaCl2 or 0.1 mM EGTA. In some experiments, Fura-2 loaded platelets were treated with 100 µM 2-MeSAMP for 5 min in the dark at 37°C prior to measuring intracellular Ca2+ mobilization. Ca2+ release from internal stores was determined by stimulating platelets with 3 µM thapsigargin. Eighty microliters of Fura-2 loaded platelets were placed in 96-well plates, stimulated with agonist, and read in a NOVOstar plate reader (BMG Labtech, Durham, NC) at 37°C. Intracellular Ca2+ variations were monitored by measuring the Fura-2 fluorescence ratio at 340/380 nm with emission at 510 nm. Fluorescence measurement was converted to the concentration of intracellular free Ca2+ using equation reported by Grynkiewicz et al. [20].
Measurement of PAR4 expression in mouse platelets
Mouse platelet surface expression of PAR4 was determined by flow cytometry using a Beckman Coulter LSRII (Case Comprehensive Cancer Center Flow Core). Washed mouse platelets were adjusted to a final concentration of 40×106 platelets/mL in HEPES-Tyrode buffer (pH 7.4). Twenty-five microliter aliquots were incubated with 25 µg/mL of anti-PAR4-FITC at 4°C for 20 min. Platelets were then diluted (1∶8) and 10,000 events were acquired on a Beckman Coulter LSRII. Flow cytometry data were analyzed with Flowjo software.
Western blotting
Washed platelets were adjusted to a final concentration of 3×108 platelets/mL. Fifty microliter aliquots were activated with thrombin(1, 10, 30, or 100 nM) or AYPGKF (0.03, 0.5, 1.5, or 2 mM) for 3 min at 37°C. The reaction was stopped by adding 6× Laemmli reducing buffer and samples were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were incubated with primary rabbit antibody to phospho-(Ser) PKC substrates, PKC, or phospho-Akt (Ser473). To demonstrate protein loading, the membrane was reprobed with a rabbit antibody to α-actinin. Detection was performed with HRP-conjugated anti-rabbit secondary antibody and an enhanced chemiluminescence system (Pierce Chemical). The optical densities of proteins in the blot were quantified using Image J (1.45) software.
ELISA-based RhoA activation assay
RhoA GTPase activity was determined in freshly prepared mouse platelets lysates (adjusted to a final concentration of 3×108 platelets/mL) by using the absorbance based G-LISA RhoA activation assay kit (Cytoskeleton, Inc.). Fifty microliter aliquots were activated with thrombin (1, 10, 30, or 100 nM) for 3 min at 37°C. Activated mouse platelets were then lysed using an equal volume of supplied cell lysis buffer, and the lysates were immediately used for the Rho G-LISA assay. All subsequent incubation and detection followed the instructions provided by the manufacturer.
Bioluminescence resonance energy transfer (BRET) assay and cell surface expression of mouse PAR3 and mouse PAR4 measurement
The cDNA for mouse PAR3 was purchased from Origene Technologies Inc. (Rockville, MD) and the cDNA for mouse PAR4 was isolated from a BaF3 cDNA library. An amino terminal HA (YPYDVPDYA) or V5 (GKPIPMPLLGLDST) epitope tag was added to PAR3 or PAR4 by PCR, and the cDNAs were ligated into pLUC-N2 (HA-tagged) or pGFP2-N2 (V5 tagged). The mouse rhodopsin in pGFP2-N2 (V5 tagged) has been described [21] . The BRET vectors were purchased from PerkinElmer. All constructs were verified by DNA sequencing. For BRET assays, HEK293 cells (American Type Culture Collection) were transfected with Lipofectamine 2000 according to manufacturer's instructions. BRET assays were performed as previously described [21].
Cell surface expression of mouse PAR3 and mouse PAR4 was determined by flow cytometry using BD LSRFortessa (Center for Aids Research, Immune Function Core, Case Western Reserve University). HEK293 cells (1×105) in 6-well plates were transfected with 1.5 µg of HA-PAR4-LUC or V5-PAR4-GFP and 1 µg of HA-PAR3-LUC or V5-PAR3-GFP. Transfected cells were removed from plates 48 h post-transfection by rinsing with PBS. Surface detection of HA- tagged PAR3 or PAR4 was detected with an HA tag antibody conjugated to Alexa Fluor 647 (Cell Signaling Technology Inc) with 1∶50 dilution. Surface detection of V5- tagged PAR3 or PAR4 was detected with a V5 tag antibody conjugated to Alexa Fluor 647 (AbD Serotec) with 1∶5 dilution. HEK293 labeled cells (1.25×105) were then diluted (1∶8) and 10,000 events were acquired on a BD LSRFortessa. Cell surface expression of HA- or V5-tagged PAR4 or PAR3 was determined by quantitative flow cytometry and performed essentially as described [21].
Data analysis
Differences between means were determined by unpaired Student's t test and by one way ANOVA test and were considered significant when p<0.05.
Results
Intracellular Ca2+ mobilization is increased in PAR3−/− mouse platelets
We first determined if the absence of PAR3 affected PAR4 mediated intracellular Ca2+ mobilization in PAR3−/− platelets in response to thrombin. The EC50 for thrombin-induced Ca2+ mobilization is increased ∼10-fold in PAR3−/− platelets compared to wild type platelets (4.1 nM vs 0.6 nM, with a 95% confidence interval of 0.24–1.5 nM or 2.3–15 nM, respectively) (Figure 1A). Heterozygous mice (PAR3+/−) had an intermediate value (1.1 nM with a 95% confidence interval of 0.5–3.7 nM). These results agree with published data showing that PAR3 is a cofactor for PAR4 activation at low thrombin concentrations [6]. However, at thrombin concentrations above 10 nM, platelets from PAR3−/− mice had a ∼1.6-fold increase in the maximum Ca2+ mobilization compared to wild type platelets. Platelets from PAR3+/− had an intermediate increase in the maximum Ca2+ mobilization (∼1.2-fold) (Figure 1A). These data indicated that the absence of PAR3 affects the Ca2+ mobilization in response to high thrombin concentrations (30–100 nM). We next determined if the increase in the maximum Ca2+ mobilization in PAR3−/− platelets was dependent on thrombin's interaction with PAR4 by using a specific PAR4 activating peptide (AYPGKF). Similar to thrombin treatment, platelets from PAR3−/− and PAR3+/− mice had a 2.6-fold or 1.9-fold increase in the maximum Ca2+ mobilization, respectively, compared to wild type platelets in response to AYPGKF (Figure 1B). The EC50 for AYPGKF-induced Ca2+ mobilization is not statistically significant between wild type and PAR3−/− platelets (392 µM vs. 614 µM, respectively, p = 0.45). Next, we investigated whether the increase in the maximum Ca2+ mobilization in the PAR3−/− mice was specific to PAR4 stimulation. Wild type and PAR3−/− platelets were stimulated with convulxin, the specific GPVI agonist, or with a high concentration of ADP [22], the specific P2Y12 and P2Y1 agonist. There was no significant difference in the maximum Ca2+ mobilization or the EC50 in response to convulxin (Figure 1C) or in the maximum Ca2+ mobilization in response to 20 µM ADP (Figure 1D) between wild type and PAR3−/− platelets. These data indicate that the increase in the maximum Ca2+ mobilization was specific to PAR activation, but independent of the PAR4 agonist. These data suggest that PAR3 influences PAR4 at the level of the receptor. To verify that the increase in the maximal Ca2+ mobilization was not due to an increase in surface expression of PAR4 in PAR3−/− platelets, PAR4 expression was measured by flow cytometry. Platelets from wild type and PAR3−/− mice had the same level of PAR4 expression (Figure 2).
Figure 1. Dose response curve of Ca2+ mobilization in the presence of extracellular Ca2+ in mouse platelets.

Fura 2-loaded wild type (black circle), PAR3−/− (gray circle), and PAR3+/− (white square) platelets were activated with the indicated concentrations of: (A) thrombin, (0.001–100 nM, (B) AYPGKF (0–2 mM), (C) convulxin (0.01–100 nM), or 20 µM of ADP for 10 min at 37°C in the presence of 2 mM of CaCl2. The difference between the maximum increase and the basal intracellular Ca2+ mobilization was measured. The results are the mean (± SD) of three independent experiments (* p<0.05).
Figure 2. PAR4 expression on mouse platelets.
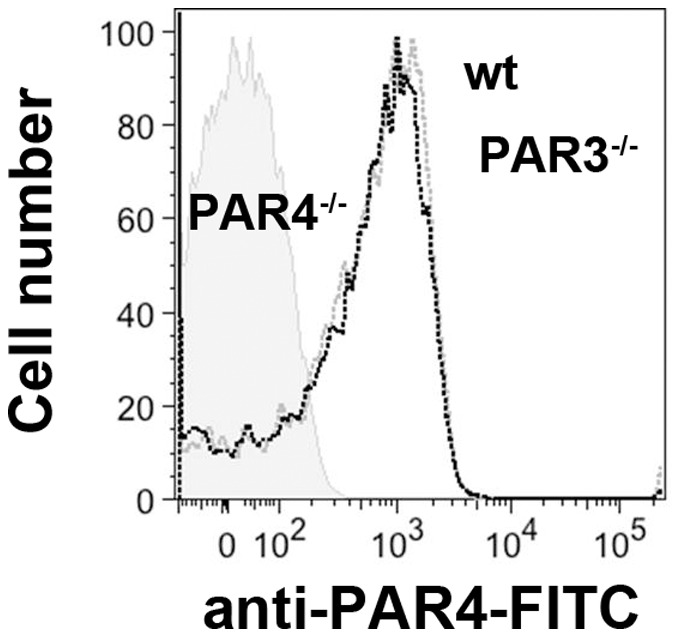
Flow cytometric analysis of PAR4 expression in wild type (WT) (black line), PAR3−/− (gray line), and PAR4−/− (shaded) mice platelets using anti-PAR4-FITC antibodies.
P2Y12 inhibition does not influence PAR4 enhanced Ca2+ mobilization in PAR3−/− mouse platelets
PAR4 and P2Y12 physically interact in human platelets after thrombin or AYPGKF stimulation and the association is reduced by P2Y12 inhibitor 2MeSAMP [23]. To determine if the increase in the maximum Ca2+ mobilization was caused by crosstalk between PAR4 and P2Y12 in the absence of PAR3, wild type and PAR3−/− platelets were stimulated with thrombin or AYPGKF in the presence of 2MeSAMP (P2Y12 antagonist). There was no significant difference in the maximum Ca2+ mobilization between wild type and PAR3−/− platelets activated with 30 nM thrombin (p = 0.64, data not shown) or 100 nM thrombin (p = 0.99, Figure 3A). Similarly, there was no significant difference in maximum Ca2+ mobilization when platelets were stimulated with 1.5 mM AYPGKF (p = 0.10, data not shown) or 2 mM AYPGKF (p = 0.06, Figure 3B). These data indicate that the increase in the maximum Ca2+ mobilization was independent of the PAR4-P2Y12 interaction after thrombin or AYPGKF stimulation.
Figure 3. Effect of 2MeSAMP on PAR4 enhancing intracellular Ca2+ mobilization in mouse platelets.

Fura 2-loaded wild type (black) and PAR3−/− (gray) platelets were incubated at 37°C for 5 min in the absence or the presence of 100 µM 2MeSAMP. After treatment, platelets were activated 100 nM thrombin (A,) or 2 mM AYPGKF (B) for 10 min at 37°C in the presence of 2 mM of CaCl2. The difference between the maximum increase and the basal intracellular Ca2+ mobilization was measured. The results are the mean (± SD) of three independent experiments (* p<0.05).
Protein Kinase C (PKC) activation is increased in PAR3−/− mouse platelets
Intracellular Ca2+ mobilization and PKC activation are both downstream of Gq. We next determined if PKC activation was also increased in PAR3−/− platelets by measuring the serine phosphorylation of PKC substrates, which reflects the activation of PKC. Serine phosphorylation on PKC substrates in response to 1 nM thrombin was absent in PAR3−/− platelets compared to wild type platelets (Figure 4A and B). These results are expected because PAR3 is required for PAR4 activation at low concentrations of thrombin. Importantly, we show that the level of serine phosphorylation of PKC substrates was increased in response to high concentration of thrombin (30–100 nM) in PAR3−/− compared to wild type mouse platelets. The level of serine phosphorylation on PKC substrates was also increased in response to AYPGKF in PAR3−/− platelets (Figure 4C and D). To show that the increased PKC activity in PAR3−/− platelets was not due to increased expression of PKC, we reprobed the membranes for total PKC. There was no significant difference in the level of PKC between PAR3−/− and wild type platelets. These results are consistent with the Ca2+ mobilization data and indicate that PAR3 negatively regulates PAR4-induced Gq-mediated signaling in mouse platelets.
Figure 4. Western blot analysis of protein kinase C (PKC) substrate phosphorylation in mouse platelets.

The level of PKC substrate phosphorylation on serine residues in response to increasing concentrations of: (A) thrombin (1–100 nM) or (C) AYPGKF (0.03–2 mM) was determined by western blotting with phospho-(Ser) PKC substrate antibody. The membranes were re-probed for α-actinin to demonstrate protein loading. The blots shown are from a representative of three independent experiments. Quantitation of PKC substrate phosphorylation in response to (B) thrombin or (D) AYPGKF is represented at the mean (± SD) (* p<0.05).
Intracellular Ca2+ store depletion is increased in PAR3−/− mouse platelets
We next examined if the increase in the maximum Ca2+ mobilization was caused by an increase in the depletion of intracellular Ca2+ stores. Platelets from PAR3−/− and wild type mice were stimulated with thrombin (1, 10, 30, or 100 nM) in Ca2+-free buffer (0.1 mM EGTA added). At 1 nM thrombin, the depletion of intracellular Ca2+ stores was decreased in PAR3−/− compared to wild type platelets. These data are consistent with PAR3 facilitating PAR4 activation at low thrombin concentrations. However, at thrombin concentrations above 10 nM, platelets from PAR3−/− release more Ca2+ from the internal stores compared to wild type platelets (Figure 5A and B). The maximum Ca2+ mobilization from the intracellular stores in PAR3−/− platelets was 405±158 nM compared to 229±47 nM for wild type platelets, p = 0.04 (Figure 5B). Similar to thrombin stimulation, PAR3−/− platelets release more Ca2+ from their internal stores compared to wild type platelets in response to AYPGKF (Figure 5C and D). However, in response to 3 µM thapsigargin, there is no difference in Ca2+ release from the internal stores between PAR3−/− and wild type platelets (Figure 5E and F). These data indicate that PAR3−/− has the same Ca2+ pool in the internal stores compared to wild type platelets. However, after activation, there is more Ca2+ released from the intracellular stores for PAR3−/− platelets compared to wild type platelets.
Figure 5. Dose response curve of Ca2+ mobilization in the absence of extracellular Ca2+ in mouse platelets.

Fura 2-loaded wild type (black line) and PAR3−/− (gray line) platelets were resuspended in Ca2+-free medium (0.1 mM EGTA was added at the time of experiment). Representative tracings are shown from platelets activated with the indicated concentrations of: (A) thrombin (1–100 nM), (C) AYPGKF (0.15–2 mM), or (E) 3 µM thapsigargin (TG). Quantitation of the change in peak Ca2+ mobilization in platelets stimulated with: (B) thrombin, (D) AYPGKF, or (F) thapsigargin. The results are the mean (± SD) of three independent experiments (* p<0.05).
G12/13 and Gi-mediated signaling are not affected in PAR3−/− mouse platelets
PAR4 couples to Gq and G12/13 in human platelets [7], [8]. We next determined if G12/13-mediated signaling was also negatively regulated by PAR3 in response to thrombin in mouse platelets. The G12/13 pathway was tested by measuring the activation of the small GTPase RhoA by a G-LISA in response to thrombin. As expected, the level of RhoA activation is decreased in PAR3−/− compared to wild type mouse platelets at low thrombin concentrations (≤10 nM) because PAR4 was unable to mediate the signaling in the absence of PAR3 (Figure 6). However, there was no significant difference in the level of RhoA activation in response to thrombin concentrations (≥30 nM) in PAR3−/− platelets compared to wild type mouse platelets. We next examined the activation of Gi pathway in response to thrombin by measuring the phosphorylation level of Akt. The activation of Akt plays an important role in platelet aggregation and secretion by negatively regulating glycogen synthase kinase 3β (GSK-3β) [24], [25]. Our data show that in response to increasing concentrations of thrombin, there was no significant difference in Akt activation between PAR3−/− and wild type mouse platelets (Figure 7A and B). These data indicate that PAR3 negatively regulates PAR4 induced Gq signaling pathways without affecting G12/13 and Gi pathways in mouse platelets.
Figure 6. RhoA activity measured by G-LISA Kit in mouse platelets.
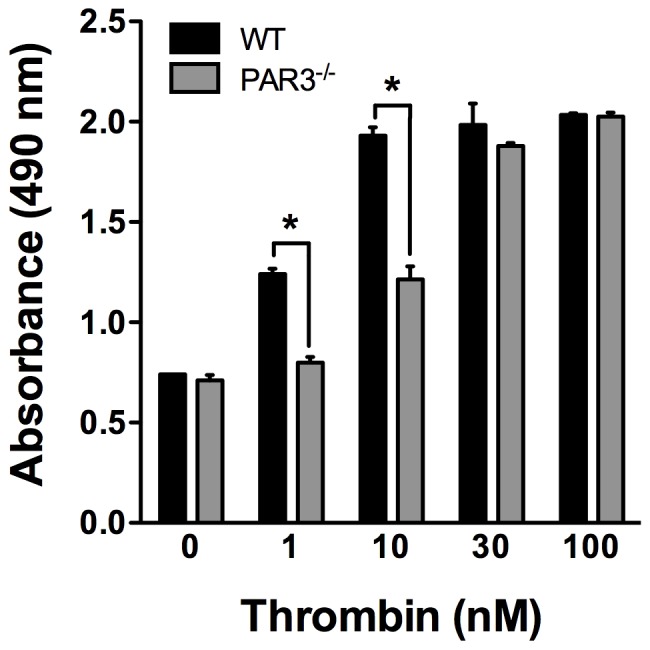
The level of activated RhoA-GTP is measured by absorbance at 490 nm in response to increasing concentrations of thrombin (1–100 nM). The results are from three independent experiments (* p<0.05, ns: not significant).
Figure 7. Western blot analysis of Akt phosphorylation in mouse platelets.
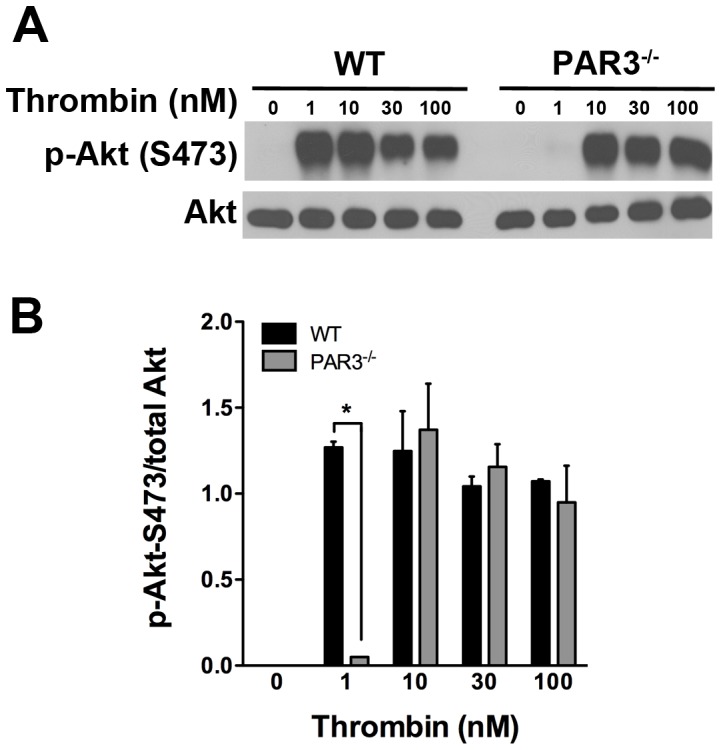
(A) The level of Akt phosphorylation at Ser473 in response to increasing concentrations of thrombin (1–100 nM) was determined by western blotting with phospho-Akt (Ser473) antibody. The membrane was re-probed for total Akt to demonstrate protein loading. The blots shown are from a representative of three independent experiments. (B) Quantitation of Akt phosphorylation at (Ser 473) in response to thrombin is represented at the mean (± SD, n = 3) (* p<0.05).
PAR3 and PAR4 form constitutive homodimers and heterodimers
To address the mechanism of how down-regulation of mouse PAR3 affects mouse PAR4 signaling, we investigated the possibility that PAR3 and PAR4 physically interact using bioluminescent resonance energy transfer (BRET) [21]. Initial studies examined the PAR3-PAR4 heterodimer (Figure 8A). PAR3 and PAR4 formed heterodimers as indicated by a hyperbolic BRET signal in response to an increase in the PAR3-GFP: PAR4-Luc ratio. We next determined that PAR3 and PAR4 also formed homodimers (Figure 8 B and C) and PAR3 or PAR4 were unable to form heterodimers with rhodopsin (Rho) (Figure 8 D and E). These data demonstrate that PAR3 specifically interact with PAR4 and form constitutive homodimers and heterodimers. Finally, we verified the expression level of PAR3 and PAR4 in HEK293 cells by flow cytometry using HA or V5 tag antibodies conjugated to Alexa Fluor 647. The mean fluorescence intensity from each antibody was converted to antibody binding sites using quantitative flow cytometry (Figure 8 F, G and H).
Figure 8. Bioluminescence resonance energy transfer (BRET) analysis of PAR3 homodimers, PAR4 homodimers, and PAR3-PAR4 heterodimers.

The HEK293 cells were transfected with: (A) PAR4-Luc (1 µg) and PAR3-GFP (0–2.5 µg), (B) PAR3-Luc (1 µg) and PAR3-GFP (0–2.5 µg), or (C) PAR4-Luc (1 µg) and PAR4-GFP (0–2.5 µg). As a control experiment, the HEK293 cells were transfected with: (D) PAR3-Luc (1 µg) and rho-GFP (0–0.12 µg), or (E) PAR4-Luc (1 µg) and rho-GFP (0–0.12 µg). Forty-eight hours post-transfection, the cells were analyzed for GFP expression, Luc expression, and BRET. The curves were plotted as the ratio of GFP to Luc and all points from 3–6 independent experiments were analyzed by global fit to a hyperbolic or linear curve. The surface expression of PAR3 and PAR4 in the HEK293 cells was determined by flow cytometry. (G) V5-PAR4-GFP and V5-PAR3-GFP were detected with a V5 tag antibody conjugated to Alexa Fluor 647. (H) HA-PAR4-LUC and HA-PAR3-LUC were detected with a HA tag antibody conjugated to Alexa Fluor 647. The results are the mean (± SD) of two independent experiments. The number of PAR4 and PAR3 molecules on the HEK293 cells surface is calculated from the V5 or HA antibody standard curve using quantitative flow cytometry (F).
Discussion
The accepted physiological role of PAR3 in mouse platelets is to serve as a cofactor for cleavage and activation of PAR4 at low thrombin concentrations [6]. The results from the current study provide the first evidence that PAR3 plays an additional role in mouse platelets by negative regulation of PAR4 mediated Ca2+ mobilization and protein kinase C (PKC) activation without affecting the downstream signaling of the G12/13 pathways. Throughout our study we have used thrombin concentrations of 30 and 100 nM. It is common to use low thrombin concentrations to examine signaling pathways in platelets so that one can detect subtle differences that would otherwise be missed. It is important to consider that the thrombin concentration generated at the platelet surface at the site of injury likely reaches >100 nM locally [19].
In human platelets, the elevation in intracellular Ca2+ concentration regulates various platelet functions, such as integrin activation, granule secretion, and rapid procoagulant phosphatidylserine (PS) exposure [26], [27]. One important initiator of Ca2+ signaling is the activation of Gq pathways, which induce the generation of diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) to activate PKC and Ca2+ store depletion, respectively [28]. In platelets, the major Ca2+ entry pathway is mediated by Ca2+ channels known as store-operated calcium entry (SOCE). The SOCE channels are activated by depletion of intracellular Ca2+ stores induced by IP3 generated downstream of Gq [29]. In this study, we have shown that platelets from PAR3−/− mice have 1.6-fold increase in the maximum intracellular Ca2+ mobilization (Figure 1), an increase in phosphorylation level of PKC substrates (Figure 4), and a 2-fold increase in Ca2+ release from the stores (Figure 5) in response to thrombin (30–100 nM) or AYPGKF. Our results from Ca2+ store depletion are consistent with previous data that show an increase in IP3 formation in COS7 cells transfected with PAR4 compared to COS7 transfected with both receptors (PAR4 and PAR3) in response to thrombin (10–100 nM) [6]. It has been shown that PAR1, but not PAR4, negatively regulates intracellular Ca2+ mobilization and procoagulant phosphatidylserine (PS) exposure in a PKC-dependent mechanism in human platelets [30]. Our data show that PAR3 negatively regulates Ca2+ mobilization and PKC activation in response to high thrombin concentration or PAR4 agonist peptide, perhaps by a physical interaction with PAR4 in mouse platelets. Further, platelets from PAR3+/− had an intermediate increase in Ca2+ mobilization (Figure 1A and B). These data support that PAR3 is directly influencing signaling from PAR4. In platelets, PAR4 also interacts with the P2Y12 receptor in response to thrombin [23]. Therefore, it is also possible that PAR4 and P2Y12 heterodimers are increased in the absence of PAR3, which influences PAR4 mediated increase in the maximum Ca2+ mobilization. However, our results show that blocking ADP signaling with 2MeSAMP does not affect the Ca2+ mobilization in response to thrombin (30 and 100 nM) or AYPGKF (1.5 and 2 mM) in PAR3−/− platelets. These data confirm that PAR3 is affecting the Ca2+ signaling downstream of PAR4 independently of P2Y12.
PAR subtypes communicate with one another to modulate signaling [14], [15], [21], [31]. It has been reported that PAR3 is able to enhance the cleavage of PAR4 with thrombin in cells expressing PAR4 and the N-terminal domain of PAR3 linked to CD8 [6]. It is unlikely that the PAR3-CD8 is dimerizing with PAR4. These data indicate that the interaction between PAR3 and PAR4 is not required for enhanced cleavage of PAR4 by thrombin. Our data suggest that PAR3's ability to enhance PAR4 cleavage is distinct from its influence on PAR4 signaling. We demonstrate in the current study that PAR3 directly interacts with PAR4 and forms constitutive homodimers and heterodimers by using bioluminescent resonance energy transfer (BRET) (Figure 8). The balance between PAR3 homodimers, PAR4 homodimers, and PAR3-PAR4 heterodimers maybe altered in the absence of PAR3 in mouse platelets, which may influence the Gq signaling pathway. It is widely accepted that PAR3 does not signal on its own. However, there are two examples of PAR3 regulating signaling from other PAR family members. McLaughlin et al. showed that the activation of PAR1-PAR3 heterodimers with thrombin induces distinct signaling from PAR1-PAR1 homodimers [17]. A second example is in podocytes where PAR3 influenced activated protein C (APC)-mediated cytoprotective signaling through PAR1 in mice or PAR2 in humans [18]. The debate between monomers, dimers, and oligomers is ongoing in the GPCR field. However, recent technological advances using sophisticated FRET detection systems have suggested that some GPCRs form parallelogram shaped tetramers [32]. In addition, one report using this technique has demonstrated an excess of dimers in addition to tetramers [33]. The authors hypothesize that there are two interfaces of the complexes with differing affinities, a high affinity site (dimers) and a lower affinity site for the tetramers (a dimer of dimers). It is quite possible that we are seeing dimers of PAR3 interact with dimers of PAR4. As discussed above, PAR4 also forms heterodimers with the P2Y12 receptor [23]. The P2Y12 receptors may be oligomerizing with dimers of PAR4 in a similar manner. At this point we are limited by the technology to detect these higher order structures in a quantitative manner in native platelet membranes. The understanding of how GPCRs cooperate physically to mediate signaling is crucial to understanding their function and should be the focus of future studies
PAR4 is also coupled to G12/13 in platelets [7]. The activation of the G12/13 pathway by thrombin induces the activation of the small GTPase RhoA which regulates dense granule release and platelet shape change [7]. Our data show that the activation level of RhoA-GTP (Figure 6) is not affected in PAR3−/− platelets compared to wild type mouse platelets in response to thrombin (30–100 nM). These results demonstrate that PAR4 signaling through Gq, but not G12/13, is regulated by PAR3. The direct coupling of PAR4 to Gi in platelets has been attributed to indirect activation of Gi pathways via secreted ADP acting on its receptors [12]. In other studies, Akt activation downstream of PARs was G12/13 and Gi dependent, but independent of Gq [8]. In our studies, we have used Akt phosphorylation as a measure of Gi activation. There were no significant differences between wild type platelets and PAR3−/− platelets (see Figure 7). There is one report that Akt phosphorylation is downstream of phospholipase C (PLC) in human platelets [34]. Reséndiz et al. showed that late Akt phosphorylation was dependent on PLC, calcium, PKC and PI3K in human platelets stimulated with thrombin (1 U/mL) or AYPGKF (0.25 mM). In our study we show significant differences in Ca2+ mobilization and PKC activation in response to thrombin concentrations (≥30 nM which corresponds to ≥4 U/mL) or AYPGKF concentrations (≥0.5 mM) in PAR3−/− compared to wild type mouse platelets. However, we do not see changes in Akt phosphorylation in our studies. It is possible that at high agonist concentrations for 3 minutes, the activation of Akt is driven primarily by Gi signaling rather than PLC. Taken together, our results show an increase Gq dependent signaling in PAR3 −/− mice.
In summary, when PAR4 is activated in the absence of PAR3 with high concentrations of thrombin (≥10 nM) or PAR4 activated peptide (≥0.5 mM), the Gq signaling pathway is increased. In order to explain the increased Ca2+ mobilization in PAR3−/− platelets, which affects the Gq-dependent signaling, but not G12/13-dependent signaling, we considered the following hypotheses. First, PAR3 can regulate Gq signaling indirectly through PAR4. The absence of PAR3 may induce a conformational change in PAR4, which increases the activity of Gq. However, PAR4 is also coupled to G12/13 and a conformational change in PAR4 would also affect the signaling downstream of G12/13. A global change in PAR4 activity by PAR3 is not consistent with our results since the G12/13 signaling pathway was not affected in PAR3−/− mice. A second hypothesis is that the expression or distribution of proteins such as RGS (Regulator of G-protein Signaling) is altered in the PAR3−/− mice. A recent study showed that preventing RGS/Spinophilin/and tyrosine phosphatase SHP-1 complex formation in platelet produced a gain in function and increase Gq-mediated signaling [35]. However, as PAR3+/− platelets produced an intermediate level of Ca2+ mobilization, it is unlikely that RGS expression, function, or both are affected in PAR3−/−. A third hypothesis is that PAR3 selectively regulates Gq signaling by direct contact with PAR4 and this is altered in PAR3−/− mice due to a change in the ratio of PAR4 homodimers to PAR3-PAR4 heterodimers. This hypothesis is supported by the study carried out on the serotonin 5-hydroxytryptamine2C (5-HT2C) receptors one of the largest subfamilies of GPCRs expressed in platelets [36]. The results of this study show that 5-HT2C homodimer interacts with a single Gq protein and the dimerization plays a functional role in regulating the activity of 5-HT2C receptors expressed in HEK293 cells. Recent studies from our laboratory have shown that human PAR4 forms constitutive homodimers at the plasma membrane [21]. Mutations at the PAR4-PAR4 homodimer interface disrupted dimer formation and reduced Ca2+ mobilization in response to the PAR4 agonist peptide [21]. PAR3-PAR4 heterodimer may have reduced capacity in comparison to the PAR4 homodimer. The increased regulation of signaling due to the physical interaction may allow for fine-tuning of responses from GPCRs. A recent study by McCoy et al. was able to differentiate the binding site for Gq and G12/13 on PAR1 [10]. The specific region on PAR4 that interacts with G-proteins is unknown. Future studies that examine these interactions may also uncover how PAR subtypes modulate PAR4 signaling. These are important considerations and potential consequences for PAR4 interactions with PAR1 on human platelets.
In conclusion, this work reports that PAR3 negatively regulates PAR4-mediated Ca2+ mobilization and PKC activation at physiologic thrombin levels by a physical interaction with PAR4 without influencing G12/13 signaling. The influence on Gq signaling is distinct from PAR3 enhancing PAR4 activation at threshold thrombin levels, as described previously by Nakanishi-Matsui et al. [6]. Given the importance of PAR4 activation in platelets of multiple species, the interactions of PAR4 with other receptors may influence downstream signaling and cellular response. Uncovering the details of these interactions is essential to fully understanding the optimal targets for future antiplatelet therapies. These studies also shed light on how PAR3 may modulate signaling in other cell types to modulate specific signaling cascades.
Acknowledgments
The authors would like to thank Michele Mumaw for helpful suggestions and editing of the manuscript.
Funding Statement
The work was supported by the American Society for Hematology (Scholar Award) and the National Institutes of Health (R01 HL098217) to MN. This research was also supported by the Cytometry and Light Microscopy Core Facility of the Comprehensive Cancer Center of Case Western Reserve University and University Hospitals of Cleveland (P30 CA43703). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vu TK, Hung DT, Wheaton VI, Coughlin SR (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64: 1057–1068. [DOI] [PubMed] [Google Scholar]
- 2. Nanevicz T, Ishii M, Wang L, Chen M, Chen J, et al. (1995) Mechanisms of thrombin receptor agonist specificity. Chimeric receptors and complementary mutations identify an agonist recognition site. J Biol Chem 270: 21619–21625. [DOI] [PubMed] [Google Scholar]
- 3. Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR (1999) Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest 103: 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ishihara H, Zeng D, Connolly AJ, Tam C, Coughlin SR (1998) Antibodies to protease-activated receptor 3 inhibit activation of mouse platelets by thrombin. Blood 91: 4152–4157. [PubMed] [Google Scholar]
- 5. Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, et al. (1998) A dual thrombin receptor system for platelet activation. Nature 394: 690–694. [DOI] [PubMed] [Google Scholar]
- 6. Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, et al. (2000) PAR3 is a cofactor for PAR4 activation by thrombin. Nature 404: 609–613. [DOI] [PubMed] [Google Scholar]
- 7. Jin J, Mao Y, Thomas D, Kim S, Daniel JL, et al. (2009) RhoA downstream of G(q) and G(12/13) pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem Pharmacol 77: 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim S, Jin J, Kunapuli SP (2006) Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood 107: 947–954. [DOI] [PubMed] [Google Scholar]
- 9. Voss B, McLaughlin JN, Holinstat M, Zent R, Hamm HE (2007) PAR1, but not PAR4, activates human platelets through a Gi/o/phosphoinositide-3 kinase signaling axis. Mol Pharmacol 71: 1399–1406. [DOI] [PubMed] [Google Scholar]
- 10. McCoy KL, Gyoneva S, Vellano CP, Smrcka AV, Traynelis SF, et al. (2012) Protease-activated receptor 1 (PAR1) coupling to G(q/11) but not to G(i/o) or G(12/13) is mediated by discrete amino acids within the receptor second intracellular loop. Cell Signal 24: 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jantzen HM, Milstone DS, Gousset L, Conley PB, Mortensen RM (2001) Impaired activation of murine platelets lacking G alpha(i2). J Clin Invest 108: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, et al. (2002) Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood 99: 3629–3636. [DOI] [PubMed] [Google Scholar]
- 13. Weiss EJ, Hamilton JR, Lease KE, Coughlin SR (2002) Protection against thrombosis in mice lacking PAR3. Blood 100: 3240–3244. [DOI] [PubMed] [Google Scholar]
- 14. Nieman MT (2008) Protease-activated receptor 4 uses anionic residues to interact with alpha-thrombin in the absence or presence of protease-activated receptor 1. Biochemistry 47: 13279–13286. [DOI] [PubMed] [Google Scholar]
- 15. Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, et al. (2006) Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation 113: 1244–1254. [DOI] [PubMed] [Google Scholar]
- 16. Mao Y, Jin J, Daniel JL, Kunapuli SP (2009) Regulation of plasmin-induced protease-activated receptor 4 activation in platelets. Platelets 20: 191–198. [DOI] [PubMed] [Google Scholar]
- 17. McLaughlin JN, Patterson MM, Malik AB (2007) Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A 104: 5662–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Madhusudhan T, Wang H, Straub BK, Grone E, Zhou Q, et al. (2012) Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood 119: 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wolberg AS, Campbell RA (2008) Thrombin generation, fibrin clot formation and hemostasis. Transfus Apher Sci 38: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450. [PubMed] [Google Scholar]
- 21. de la Fuente M, Noble DN, Verma S, Nieman MT (2012) Mapping the human protease activated receptor 4 (PAR4) homodimer interface to transmembrane helix 4. J Biol Chem 287: 10414–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Daniel JL, Dangelmaier C, Jin J, Ashby B, Smith JB, et al. (1998) Molecular basis for ADP-induced platelet activation. I. Evidence for three distinct ADP receptors on human platelets. J Biol Chem 273: 2024–2029. [DOI] [PubMed] [Google Scholar]
- 23. Li D, D'Angelo L, Chavez M, Woulfe DS (2011) Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J Biol Chem 286: 3805–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li D, August S, Woulfe DS (2008) GSK3beta is a negative regulator of platelet function and thrombosis. Blood 111: 3522–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Brien KA, Stojanovic-Terpo A, Hay N, Du X (2011) An important role for Akt3 in platelet activation and thrombosis. Blood 118: 4215–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bachelot-Loza C, Badol P, Brohard-Bohn B, Fraiz N, Cano E, et al. (2006) Differential regulation of platelet aggregation and aminophospholipid exposure by calpain. Br J Haematol 133: 419–426. [DOI] [PubMed] [Google Scholar]
- 27. Arachiche A, Kerbiriou-Nabias D, Garcin I, Letellier T, Dachary-Prigent J (2009) Rapid procoagulant phosphatidylserine exposure relies on high cytosolic calcium rather than on mitochondrial depolarization. Arterioscler Thromb Vasc Biol 29: 1883–1889. [DOI] [PubMed] [Google Scholar]
- 28. Jardin I, Ben Amor N, Bartegi A, Pariente JA, Salido GM, et al. (2007) Differential involvement of thrombin receptors in Ca2+ release from two different intracellular stores in human platelets. Biochem J 401: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosado JA (2011) Acidic Ca(2+) stores in platelets. Cell Calcium 50: 168–174. [DOI] [PubMed] [Google Scholar]
- 30. Harper MT, Poole AW (2011) PKC inhibition markedly enhances Ca(2+) signalling and phosphatidylserine exposure downstream of protease-activated receptor-1 but not protease-activated receptor-4 in human platelets. J Thromb Haemost 9: 1599–1607. [DOI] [PubMed] [Google Scholar]
- 31. Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, et al. (2007) ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol 8: 1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raicu V, Stoneman MR, Fung R, Melnichuk M, Jansma DB, et al. (2009) Determination of supramolecular structure and spatial distribution of protein complexes in living cells. Nature Photonics 3: 107–113. [Google Scholar]
- 33. Pisterzi LF, Jansma DB, Georgiou J, Woodside MJ, Chou JT, et al. (2010) Oligomeric size of the m2 muscarinic receptor in live cells as determined by quantitative fluorescence resonance energy transfer. J Biol Chem 285: 16723–16738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Resendiz JC, Kroll MH, Lassila R (2007) Protease-activated receptor-induced Akt activation–regulation and possible function. J Thromb Haemost 5: 2484–2493. [DOI] [PubMed] [Google Scholar]
- 35. Ma P, Cierniewska A, Signarvic R, Cieslak M, Kong H, et al. (2012) A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood 119: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herrick-Davis K, Grinde E, Harrigan TJ, Mazurkiewicz JE (2005) Inhibition of serotonin 5-hydroxytryptamine2c receptor function through heterodimerization: receptor dimers bind two molecules of ligand and one G-protein. J Biol Chem 280: 40144–40151. [DOI] [PubMed] [Google Scholar]