

Abstract
Background
Cellular respiration is the process by which cells obtain energy from glucose and is a very important biological process in living cell. As cells do cellular respiration, they need a pathway to store and transport electrons, the electron transport chain. The function of the electron transport chain is to produce a trans-membrane proton electrochemical gradient as a result of oxidation–reduction reactions. In these oxidation–reduction reactions in electron transport chains, metal ions play very important role as electron donor and acceptor. For example, Fe ions are in complex I and complex II, and Cu ions are in complex IV. Therefore, to identify metal-binding sites in electron transporters is an important issue in helping biologists better understand the workings of the electron transport chain.
Methods
We propose a method based on Position Specific Scoring Matrix (PSSM) profiles and significant amino acid pairs to identify metal-binding residues in electron transport proteins.
Results
We have selected a non-redundant set of 55 metal-binding electron transport proteins as our dataset. The proposed method can predict metal-binding sites in electron transport proteins with an average 10-fold cross-validation accuracy of 93.2% and 93.1% for metal-binding cysteine and histidine, respectively. Compared with the general metal-binding predictor from A. Passerini et al., the proposed method can improve over 9% of sensitivity, and 14% specificity on the independent dataset in identifying metal-binding cysteines. The proposed method can also improve almost 76% sensitivity with same specificity in metal-binding histidine, and MCC is also improved from 0.28 to 0.88.
Conclusions
We have developed a novel approach based on PSSM profiles and significant amino acid pairs for identifying metal-binding sites from electron transport proteins. The proposed approach achieved a significant improvement with independent test set of metal-binding electron transport proteins.
Introduction
Cellular respiration is the process by which cells obtain energy from glucose. During respiration, cells break down simple food molecules, such as sugar, and release the energy they contain [1]. The point of cellular respiration is to harvest electrons from organic compounds such as glucose and use that energy to make a molecule called ATP (adenosine triphosphate). ATP in turn is used to provide energy for most cellular reactions.
As cells do cellular respiration, they need a pathway to store and transport electrons, the electron transport chain. The function of the electron transport chain is to produce a trans-membrane proton electrochemical gradient as a result of oxidation-reduction reactions. If protons flow back through the membrane, ATP synthase converts this mechanical into chemical energy by producing ATP, which is provided energy in many cellular processes. The architecture of the electron transport chain with complex I–IV is given in Figure 1.
Figure 1. The electron transport chain in the inner membrane of mitochondria.

As Figure 1 shows, at the mitochondrial inner membrane, electrons from NADH and succinate pass through the electron transport chain to oxygen (Complex I(NADH dehydrogenase) and Complex II(succinate dehydrogenase)). Electrons pass from complex I to a carrier (coenzyme Q) embedded by itself in the membrane. From coenzyme Q electrons are passed to a Complex III (cytochrome b, c1 complex). Note that the path of electrons is from Complex I to Coenzyme Q to Complex III. Complex II, the succinate dehydrogenase complex, is a separate starting point, and is not a part of the NADH pathway. From Complex III the pathway is to cytochrome c then to a Complex IV (cytochrome oxidase complex). In the end, the proton electrochemical gradient allows ATP synthase to use the flow of H+ to generate ATP.
There are many oxidation-reduction reactions in the electron transport chain, such as NADH dehydrogenase, coenzyme Q – cytochrome c reductase, and succinate – coenzyme Q reductase. In these oxidation-reduction reactions in electron transport chains, metal ions play very important role as electron donor and acceptor. For example, Fe ions are in complex I and complex II, and Cu ions are in complex IV. Therefore, to identify metal-binding sites in electron transporters is an important issue in helping biologists better understand the workings of the electron transport chain. In this work, we try to develop a method based on Position Specific Scoring Matrix (PSSM) profiles and significant amino acid pairs to identify metal-binding residues in electron transport proteins.
In recent years, several methods have been proposed for predicting metal-binding sites (MBS) in proteins based on neural networks and support vector machines [2]–[5]. These work are major from A. Passerini and his co-workers except the work from Lin [2]. Prof. Passerini has proposed a two-stage machine-learning approach on their work [4]. The first stage consists of a support vector machine classifier, and the second stage consists of a bidirectional recurrent neural network. The authors of the work [4] have also published their web server as MetalDetector [5], which is the most popular web server for prediction metal-binding sites in proteins.
According to a recent comprehensive review 6], to establish a really useful statistical predictor for a protein system, we need to consider the following procedures: (i) construct or select a valid benchmark dataset to train and test the predictor; (ii)formulate the protein samples with an effective mathematical expression that can truly reflect their intrinsic correlation with the attribute to be predicted; (iii) introduce or develop a powerful engine to operate the prediction; (iv) properly perform cross-validation tests to objectively evaluate the anticipated accuracy of the predictor; (v) establish a user-friendly web-server for the predictor that is accessible to the public.
In this work, we propose a method based on PSSM profiles and significant amino acid pairs to identify metal-binding residues in electron transport proteins. We have selected a non-redundant set of 55 metal-binding electron transport proteins as our dataset. The proposed method can predict metal-binding sites in electron transport proteins with an average 10-fold cross-validation accuracy of 93.2% and 93.1% for metal-binding cysteine and histidine, respectively. Comparing with the general metal-binding predictor from A. Passerini et al., the proposed method can improve over 9% of sensitivity, and 14% specificity on the independent dataset in identifying metal-binding cysteines. The proposed method can also improve almost 76% sensitivity with same specificity in metal-binding histidine, and MCC is also improved from 0.28 to 0.88. The proposed approach achieved a significant improvement with independent test set of metal-binding electron transport proteins.
Materials and Methods
This work focuses on identifying metal-binding sites efficiently in electron transport proteins. As Figure 2 shows, the analyzing flowchart includes three sub-processes: data collection, feature set generation, and model evaluation. Following this model, we have developed a novel approach based on PSSM profiles and significant amino acid pairs for identifying metal-binding sites from electron transport proteins. The details of the proposed approach are described as follows.
Figure 2. The flowchart of ETMB-RBF.

Data collection
First of all, we selected electron transport proteins with metal binding sites from UniProt database [7]. Then, we removed the sequences without the evidence at protein level and experimental metal-binding sites. Next, by using BLAST [8], the sequences with sequence identity more than 20% were excluded from the dataset. Since sequences falling below a 20% sequence identity can have very different structure [9], it is difficult to get a high success rate when tested by dataset in excluding homologous sequences with 20% sequence identity. Finally, 55 electron transport proteins are surveyed in this work.
The collected electron transport protein sequences were divided into two datasets: the training dataset and the independent test dataset. The training dataset is used for identifying metal binding sites and evaluating significant amino acid pairs in electron transport proteins. The training dataset includes 44 electron transport protein sequences which contain 79 metal-binding cysteine, 77 metal-binding histidine and 368 non-metal-binding cysteine and histidine. The independent test dataset, which includes 11 electron transport proteins which contain 22 metal-binding cysteine, 21 metal-binding histidine and 103 non-metal-binding cysteine and histidine, is used to evaluate the performance of the proposed method. The details of two datasets are listed in Table 1 and Table 2. Table 3 summarizes the statistics of structural topology and molecular function on 55 electron transporters in this work.
Table 1. The statistic of experimentally verified metal binding sites on electron transporters.
| Cross-Validation Dataset | Independent Test Dataset | |
| Number of electron transporters | 44 | 11 |
| Number of metal binding cysteine | 79 | 22 |
| Number of metal binding histidine | 77 | 21 |
| Number of non-metal binding cysteine and histidine | 368 | 103 |
Table 2. The catalytic of experimentally verified metal binding sites on electron transporters.
| Cross-Validation Dataset | Independent Test Dataset | |||
| Cysteine | Histidine | Cysteine | Histidine | |
| Number of iron binding sites | 72 | 63 | 18 | 15 |
| Number of copper binding sites | 7 | 14 | 4 | 6 |
Table 3. Details of electron transporters in the present study.
| UniProt ID | Sequence length | Protein name | Num. of TM segment | Source | Molecular Function |
| Q5SJ79 | 562 | Cytochrome c oxidase subunit 1 | 13 | Thermus thermophilus | electron carrier activity;cytochrome-c oxidase activity; |
| P98002 | 558 | Cytochrome c oxidase subunit 1-beta | 12 | Paracoccus denitrificans | electron carrier activity;cytochrome-c oxidase activity; |
| P0A405 | 755 | PsaA | 11 | Thermosynechococcus elongatus | chlorophyll binding; |
| P51131 | 687 | Cytochrome b/c1 | 10 | Bradyrhizobium japonicum | electron carrier activity;oxidoreductase activity; |
| Q02761 | 445 | Cytochrome b | 8 | Rhodobacter sphaeroides | Electron carrier activity;Oxidoreductase activity; |
| P32791 | 686 | Ferric-chelate reductase 1 | 7 | Saccharomyces cerevisiae | electron carrier activity;flavin adenine dinucleotide binding; |
| P0ABJ9 | 522 | Cytochrome bd-I oxidase subunit I | 7 | Escherichia coli | electron carrier activity;oxidoreductase activity; |
| P06010 | 324 | Reaction center protein M chain | 5 | Rhodopseudomonas viridis | electron transporter; |
| P0C0Y8 | 282 | Reaction center protein L chain | 5 | Rhodobacter sphaeroides | electron transporter; |
| P0A444 | 360 | Photosystem Q(B) protein 1 | 5 | Thermosynechococcus elongatus | electron transporter;oxidoreductase activity; |
| P11695 | 311 | Reaction center protein L chain | 5 | Chloroflexus aurantiacus | electron transporter;bacteriochlorophyll binding; |
| P17413 | 256 | Fumarate reductase cytochrome b subunit | 5 | Wolinella succinogenes | oxidoreductase activity; |
| P11350 | 225 | Cytochrome B-NR | 5 | Escherichia coli | electron carrier activity;nitrate reductase activity; |
| P00165 | 215 | Cytochrome b6 | 4 | Spinacia oleracea | electron transporter;oxidoreductase activity; |
| P0AEK7 | 217 | FDH-N subunit gamma | 4 | Escherichia coli | electron carrier activity;formate dehydrogenase (NAD+) activity; |
| A5GZW8 | 159 | CybS | 3 | Sus scrofa | ubiquinone binding; |
| D0VWV4 | 169 | CYBL | 3 | Sus scrofa | electron carrier activity;succinate dehydrogenase activity; |
| P69054 | 129 | Cytochrome b-556 | 3 | Escherichia coli | electron carrier activity;succinate dehydrogenase activity;ubiquinone binding; |
| P0AC44 | 115 | Succinate dehydrogenase hydrophobic membrane anchor subunit | 3 | Escherichia coli | electron carrier activity;succinate dehydrogenase activity; |
| P08306 | 298 | Cytochrome c oxidase subunit 2 | 2 | Paracoccus denitrificans | electron carrier activity;cytochrome-c oxidase activity; |
| P68530 | 227 | Cytochrome c oxidase subunit 2 | 2 | Bos taurus | electron carrier activity;cytochrome-c oxidase activity; |
| P00167 | 134 | Cytochrome b5 | 1 | Human | Aldo-keto reductase (NADP) activity;Cytochrome-c oxidase activity; |
| P95522 | 338 | Apocytochrome f | 1 | Phormidium laminosum | electron carrier activity; |
| P00125 | 325 | Cytochrome b-c1 complex subunit 4 | 1 | Bos taurus | electron carrier activity; |
| Q02760 | 285 | Cytochrome c1 | 1 | Rhodobacter sphaeroides | electron carrier activity; |
| P04166 | 146 | Cytochrome b5 type B | 1 | Rattus norvegicus | electron transporter;enzyme activator activity; |
| Q8DIP0 | 84 | Cytochrome b559 subunit alpha | 1 | Thermosynechococcus elongatus | heme binding; |
| P95673 | 46 | Light-harvesting protein B-800/850 beta 1 chain | 1 | Rhodospirillum molischianum | electron transporter; |
| Q8DIN9 | 45 | Cytochrome b559 subunit beta | 1 | Thermosynechococcus elongatus | heme binding; |
| P07143 | 309 | Cytochrome b-c1 complex subunit 4 | 1 | Saccharomyces cerevisiae | electron transporter; |
| P0AAJ3 | 294 | FDH-N subunit beta | 1 | Escherichia coli | electron carrier activity;formate dehydrogenase (NAD+) activity; |
| P20114 | 243 | Cytochrome c1, heme protein | 1 | Euglena gracilis | electron carrier activity; |
| P08980 | 230 | Rieske iron-sulfur protein | 1 | Spinacia oleracea | electron transporter;plastoquinol-plastocyanin reductase activity;ubiquinol-cytochrome-c reductase activity; |
| P49728 | 206 | Rieske iron-sulfur protein | 1 | Chlamydomonas reinhardtii | plastoquinol-plastocyanin reductase activity;ubiquinol-cytochrome-c reductase activity; |
| P0CY48 | 191 | Rieske iron-sulfur protein | 1 | Rhodobacter capsulatus | ubiquinol-cytochrome-c reductase activity; |
| P83794 | 179 | Rieske iron-sulfur protein | 1 | Mastigocladus laminosus | electron transporter;plastoquinol-plastocyanin reductase activity;ubiquinol-cytochrome-c reductase activity; |
| Q5SJ80 | 168 | Cytochrome c oxidase subunit 2 | 1 | Thermus thermophilus | cytochrome-c oxidase activity; |
| P26789 | 53 | Light-harvesting protein B-800/850 alpha chain | 1 | Rhodopseudomonas acidophila | electron transporter;bacteriochlorophyll binding; |
| P0C0Y1 | 49 | Light-harvesting protein B-875 beta chain | 1 | Rhodobacter sphaeroides | electron transporter;bacteriochlorophyll binding; |
| P0A411 | 81 | Photosystem I iron-sulfur center | Unknown | Anabaena variabilis | electron carrier activity; |
| Q7SIB8 | 102 | Plastocyanin | Unknown | Dryopteris crassirhizoma | electron carrier activity; |
| P00289 | 168 | Plastocyanin, chloroplastic | Unknown | Spinacia oleracea | electron carrier activity; |
| Q9YHT2 | 290 | Iron-sulfur subunit of complex II | Unknown | Gallus gallus | electron carrier activity;succinate dehydrogenase activity; |
| P0A386 | 163 | Cytochrome c-550 | Unknown | Thermosynechococcus elongatus | electron carrier activity; |
| P09152 | 1247 | Nitrate reductase A subunit alpha | Unknown | Escherichia coli | electron carrier activity;oxidoreductase activity; |
| Q56223 | 783 | NADH-quinone oxidoreductase subunit 3 | Unknown | Thermus thermophilus | electron carrier activity;NADH dehydrogenase (ubiquinone) activity; |
| P11349 | 512 | Respiratory nitrate reductase 1 beta chain | Unknown | Escherichia coli | electron carrier activity;nitrate reductase activity; |
| P07173 | 356 | Cytochrome c558/c559 | Unknown | Rhodopseudomonas viridis | electron carrier activity; |
| P13272 | 274 | Cytochrome b-c1 complex subunit 5 | Unknown | Bos taurus | ubiquinol-cytochrome-c reductase activity; |
| P07014 | 238 | Succinate dehydrogenase iron-sulfur subunit | Unknown | Escherichia coli | electron carrier activity;succinate dehydrogenase activity; |
| P27197 | 235 | Auracyanin-B | Unknown | Chloroflexus aurantiacus | electron carrier activity; |
| Q8RMH6 | 162 | Auracyanin-A | Unknown | Chloroflexus aurantiacus | electron carrier activity; |
| P18068 | 145 | Plastocyanin, chloroplastic | Unknown | Chlamydomonas reinhardtii | electron carrier activity; |
| P82603 | 129 | Cytochrome c-550 | Unknown | Spirulina maxima | electron carrier activity; |
| Q56247 | 111 | Cytochrome c-551 | Unknown | Bacillus PS3 | electron carrier activity; |
Feature set generation
Position Specific Scoring Matrix Profiles
In the structural point of view, several amino acid residues can be mutated without altering the structure of a protein, and it is possible that two proteins have similar structures with different amino acid compositions. Hence, the Position Specific Scoring Matrix (PSSM) profile is adopted, which have been widely used in protein secondary structure prediction, subcellular localization, classification of transporters, prediction of transport targets and other bioinformatics problems with significant improvement [10]–[20]. The PSSM profiles are obtained by using PSI-BLAST and non-redundant (NR) protein database.
PSSM profiles can be a useful feature set to represent evolutionary information in protein sequences [11], [21]. Life on Earth originated and then evolved from a common ancestor approximately 3.7 billion years ago, sequences are more similar among species that share a more recent common ancestor, and can be used to reconstruct evolutionary histories. In this work, we searched a very large sequences database (NR database) by using PSI-BLAST to find similar sequences of the target sequence. Then, we adopted the evolutionary information contained in PSSM profiles as input to radial basis function networks.
In the identification of metal binding sites on electron transport proteins, the generated PSSM profiles contained the probability of occurrence of each type of amino acid residues at each position. Each element in PSSM profile is scaled by 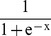 for normalizing the values between 0 and 1. The window size of 13 residues which the central residue is metal-binding site and 6 residues along both sides of the central residue is used for encapsulating an amino acid residue. Finally, 13 X 20 elements are used as PSSM feature set for identifying metal-binding sites. Features of non-metal-binding sites are generated by using the same approach as features of metal-binding sites.
for normalizing the values between 0 and 1. The window size of 13 residues which the central residue is metal-binding site and 6 residues along both sides of the central residue is used for encapsulating an amino acid residue. Finally, 13 X 20 elements are used as PSSM feature set for identifying metal-binding sites. Features of non-metal-binding sites are generated by using the same approach as features of metal-binding sites.
In addition, we also generated different feature sets for comparison. There feature sets are generated by amino acid types(AA), BLOSUM62 matrix (BLOcks of Amino Acid SUbstitution Matrix) [22], and PAM250 matrix [23]. A matrix of 13 X 20 elements is used to represent each residue in a training dataset, where 13 denotes the window size and 20 elements from each row of the type of amino acids, BLOSUM62 matrix and PAM250 matrix.
Significant amino acid pairs
The significant amino acid pairs (SAAPs) around the metal-binding sites are identified based on the training dataset. These SAAPs are adopted to construct learning model for improving performance [24]. In order to make further investigations of substrate sites specificity, these SAAPs are identified based on statistical measurement of hypergeometric distribution. Each amino acid pairs surrounding metal-binding site is calculated p-value of hypergeometric distribution. The hypergeometric distribution is defined as:
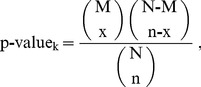 |
(1) |
where N denotes the number of sequences in the whole dataset, M denotes the number of sequences in the positive dataset, and (N-M) denotes the number of sequences in the negative dataset; n, x and n-x denotes the number of sequences which include the k-th SAAP in the whole dataset, in the positive dataset,and in the negative dataset respectively.
The amino acid pair surrounding metal-binding sites is significant when p-value is less than the significance level. It indicates that central residue is the metal-binding site with higher probability if significant amino acid pairs appear. As shown in Table 4, the most significant amino acid pair on cysteine is (−4C, 1P). (−4C, 1P), which suggests that the cysteine(C) on position −4 and the proline(P) on position +1 surrounding metal-binding sites is significant with p-value 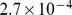 . The illustration of calculating p-value for identifying significant amino acid pairs was shown in Figure 3.
. The illustration of calculating p-value for identifying significant amino acid pairs was shown in Figure 3.
Table 4. The significant amino acid pairs that surround the metal binding cysteine and histidine on electron transporters.
| Metal binding cysteine | Metal binding histidine | ||
| SAAP | p-value | SAAP | p-value |
| (−4C,1P) | 2.70E-04 | (−4C,−1C) | 2.40E-06 |
| (4C,5P) | 2.70E-04 | (−3C,2S) | 6.00E-04 |
| (−6C,4C) | 6.90E-04 | (−3V,4V) | 6.00E-04 |
| (−3C,3C) | 1.80E-03 | (−1C,1G) | 7.20E-04 |
| (−3C,4C) | 1.80E-03 | (1G,2S) | 7.20E-04 |
| (3C,6C) | 1.80E-03 | (−3A, −1C) | 7.20E-04 |
| (−6C, −3C) | 1.80E-03 | (−1C,4Y) | 2.70E-03 |
| (−6L,3C) | 1.80E-03 | (−1L,2F) | 2.70E-03 |
| (1I,6C) | 4.50E-03 | (1S,3D) | 2.70E-03 |
| (2G,6C) | 4.50E-03 | (−2A, −1C) | 2.70E-03 |
| (−2I,3C) | 4.50E-03 | (−2I,2F) | 2.70E-03 |
| (−4G,4C) | 4.50E-03 | (−2L,3M) | 2.70E-03 |
| (−5I, −3C) | 4.50E-03 | (−2P, −1C) | 2.70E-03 |
| (−6C, −4G) | 4.50E-03 | (−2V,1S) | 2.70E-03 |
| (−6C, −5I) | 4.50E-03 | (−3D, −2V) | 2.70E-03 |
| (−6C,5P) | 4.50E-03 | (−5F, −3C) | 2.70E-03 |
| (1I,3C) | 8.60E-03 | (−5V, −1C) | 2.70E-03 |
| (−3C, −2I) | 8.60E-03 | (−5V, −3A) | 2.70E-03 |
| (1P,3G) | 1.10E-02 | (−6E, −4C) | 2.70E-03 |
| (−5I,4C) | 1.10E-02 | (−2L, −1I) | 2.80E-03 |
| (−6G, −4Y) | 1.10E-02 | (−3L,5I) | 2.80E-03 |
| (−1G,3C) | 1.90E-02 | (−4C, −3A) | 2.80E-03 |
| (−3C,1H) | 1.90E-02 | (−5F,4G) | 2.80E-03 |
| (3C,4H) | 1.90E-02 | (−5F,5M) | 2.80E-03 |
| (−4G, −3C) | 1.90E-02 | (4G,5I) | 7.60E-03 |
Figure 3. The illustration of calculating p-value for identifying significant amino acid pairs.

After calculating p-value for each amino acid pair surrounding metal-binding sites, the ranked SAAPs added into the feature set by using forward feature selection based on 10-fold cross-validation for improving predictive performance. Finally, 25 and 90 SAAPs are added into feature set of identifying metal binding cysteine and histidine, respectively. The final model was evaluated by using the independent dataset of 11 electron transporters.
The topmost 25 of SAAPs with p-value surrounding metal-binding cysteine and histidine are listed respectively in Table 4.
Model evaluation
Design of the Radial Basis Function Networks
We have employed the QuickRBF package [25] to construct RBFN classifiers in this work. Also, the fixed bandwidth of 5 for each kernel function is employed in the network. In addition, we used all training data as centers. Then, the RBFN classifier identifies metal-binding sites based on the output function value. The details about network structure and design have been explained in our earlier article [26].
Classification based on radial basis function (RBF) networks has several applications in bioinformatics. It has been widely used to predict the cleavage sites in proteins [27], inter-residue contacts [28], protein disorder [29], the discrimination of β-barrel proteins [13], the classification of transporters [14], [16],the identification of O-linked glycosylation sites [24] , and the identification of ubiquitin conjugation sites [30].
The general mathematical form of the output nodes in an RBFN is as follows:
| (2) |
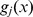 is the function corresponding to the j-th output node and is a linear combination of k radial basis functions
is the function corresponding to the j-th output node and is a linear combination of k radial basis functions 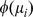 with center
with center 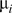 and bandwidth
and bandwidth 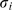 ; Also,
; Also, 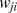 denotes the weight associated with the correlation between the j-th output node.
denotes the weight associated with the correlation between the j-th output node.
Assessment of predictive ability
The prediction performance was examined by 10-fold cross validation test, in which the training data were randomly divided into ten subsets of approximately equal size. The data were trained with nine subsets and the remaining set was used to test the performance of the method. This process was repeated 10 times so that every subset had been used as the test data once.
Sensitivity, specificity, accuracy, and MCC (Matthew's correlation coefficient) were used to measure the prediction performance. TP, FP, TN, FN are true positives, false positives, true negatives, and false negatives, respectively.
| (3) |
| (4) |
| (5) |
| (6) |
Results and Discussion
Predictive performance of metal-binding sites identification in electron transport proteins with different feature sets
We developed a variety of methods for metal-binding sites identification in electron transport proteins. The results obtained from the AA, BLOSUM62, PAM250, PSSM, and the combination of PSSM and SAAPs are presented in Table 5. The results showed that PSSM with SAAPs properties was successful in identifying metal-binding sites with an average 10-fold cross-validation accuracy of 93.2% and 93.1% for metal-binding cysteine and histidine, respectively. Our analysis showed that PSSM profiles and SAAPs properties had marginally improved the accuracy of identification, compared with the other feature sets.
Table 5. The ten-fold cross-validation performance of metal binding sites on Cross-Validation dataset.
| ETMB-RBF with different features | True Positive | False Positive | True Negative | False Negative | Sensitivity | Precision | Specificity | Accuracy | MCC |
| Metal binding cysteine | |||||||||
| AA | 60 | 22 | 90 | 19 | 75.9% | 73.2% | 80.4% | 78.5% | 0.56 |
| BLOSUM62 | 65 | 12 | 100 | 14 | 82.3% | 84.4% | 89.3% | 86.4% | 0.72 |
| PAM250 | 58 | 9 | 103 | 21 | 73.4% | 86.8% | 92.0% | 84.3% | 0.67 |
| PSSM | 76 | 16 | 96 | 3 | 96.2% | 82.6% | 85.7% | 90.1% | 0.81 |
| PSSM+SAAPs | 78 | 12 | 100 | 1 | 98.7% | 86.7% | 89.3% | 93.2% | 0.87 |
| Metal binding histidine | |||||||||
| AA | 37 | 30 | 226 | 40 | 48.1% | 55.2% | 88.3% | 79.0% | 0.38 |
| BLOSUM62 | 43 | 21 | 235 | 34 | 55.8% | 67.2% | 91.8% | 83.5% | 0.51 |
| PAM250 | 39 | 37 | 219 | 38 | 50.6% | 51.3% | 85.5% | 77.5% | 0.36 |
| PSSM | 60 | 13 | 243 | 17 | 77.9% | 82.2% | 94.9% | 91.0% | 0.74 |
| PSSM+SAAPs | 62 | 8 | 248 | 15 | 80.5% | 88.6% | 96.9% | 93.1% | 0.80 |
Combining the significant amino acid pairs with the sequence of amino acids increases the predictive accuracy specificity for metal-binding sites identification from 90.1% to 93.2% with metal-binding cysteine, and from 91.0% to 93.1% with metal-binding histidine. In addition, the sensitivity, precision specificity, and MCC are also improved. Consequently, according to the evaluation of 10-fold cross validation, the identified significant amino acid pairs can increase the predictive performance.
In statistical prediction, the following three cross-validation methods are often used to examine a prediction: independent dataset test, subsampling test, and jackknife test [6]. However, of the three test methods, the jackknife test is deemed the least arbitrary that can always yield a unique result for a given benchmark dataset. However, to reduce the computational time, we adopted the 10-fold cross validation and independent testing dataset test in this study.
Comparison the performance with other method with independent test set
The independent test dataset, which includes 11 electron transport proteins which contain 22 metal-binding cysteine, 21 metal-binding histidine and 103 non-metal-binding cysteine and histidine, is used to evaluate the performance of the proposed method. As Table 6 shows, comparing with the general metal-binding predictor from A. Passerini et al., the proposed method can improve over 9% of sensitivity, and 14% specificity on the independent dataset in identifying metal-binding cysteines. The proposed method can also improve almost 76% sensitivity with same specificity in metal-binding histidine, and MCC is also improved from 0.28 to 0.88. This results shows that our method could be effectively used for indentifying metal-binding sites in electron transport proteins.
Table 6. Comparison performance with other methods.
| Metal binding cysteine | |||||||||
| Method | True Positive | False Positive | True Negative | False Negative | Sensitivity | Precision | Specificity | Accuracy | MCC |
| Cross-Validation Dataset | |||||||||
| Metal Detector | 78 | 26 | 86 | 1 | 98.7% | 75.0% | 76.8% | 85.9% | 0.75 |
| ETMB-RBF | 78 | 12 | 100 | 1 | 98.7% | 86.7% | 89.3% | 93.2% | 0.87 |
| Independent Test Dataset | |||||||||
| Metal Detector | 20 | 8 | 21 | 2 | 90.9% | 71.4% | 72.4% | 80.4% | 0.63 |
| ETMB-RBF | 22 | 4 | 25 | 0 | 100% | 84.6% | 86.2% | 92.3% | 0.85 |
The statistical analysis of amino acid compositions in electron transporters and general proteins
We have analyzed metal-binding cysteine and cysteine residues on electron transporters and general proteins. Using the sequences of electron transporters in Table 3, we generated the sequence logos of metal-binding cysteine and cysteine residues in electron transporters with flanking amino acids (−6 ∼ +6) by WebLogo [31], [32]. Also, we generated the sequence logos of metal-binding cysteine and cysteine residues in general proteins with the dataset in A. Passerini's work [4]. These four sequence logos are listed in Figure 4.
Figure 4. The sequence frequency logos of metal-binding cysteine and histidine in electron transporters and general proteins.

We also statistically analyzed the amino acid compositions with standard T-test of metal-binding cysteine and cysteine residues in electron transporters and general proteins. As Figure 5 shows, seven residues, Q, Y, K, M, I, D and G, surrounding metal-binding cysteine have significant difference between electron transporters and general proteins. Also, 8 residues, H, K, D, E, V, Q, F, and R, surrounding metal-binding histidine have significant difference between electron transporters and general proteins.
Figure 5. The significant amino acid compositions that surround the metal binding cysteine and histidine.

According the statistical analysis, the distribution of amino acids surrounding metal-binding residues are different between electron transport proteins and general proteins. This may be the reason why our proposed method performs better than the general metal-binding predictor.
Conclusions
Cellular respiration is the process by which cells obtain energy from glucose, and is a very important biological process in living cell. As cells do cellular respiration, they need a pathway to store and transport electrons, the electron transport chain. The function of the electron transport chain is to produce a trans-membrane proton electrochemical gradient as a result of oxidation-reduction reactions. In these oxidation-reduction reactions in electron transport chains, metal ions play very important role as electron donor and acceptor. Therefore, to identify metal-binding sites in electron transporters is an important issue in helping biologists better understand the workings of the electron transport chain.
In this work, we proposed a method based on PSSM profiles and significant amino acid pairs to identify metal-binding residues in electron transport proteins. We have selected a non-redundant set of 55 metal-binding electron transport proteins as our dataset. The proposed method can predict metal-binding sites in electron transport proteins with an average 10-fold cross-validation accuracy of 93.2% and 93.1% for metal-binding cysteine and histidine, respectively. Comparing with the general metal-binding predictor from A. Passerini et al., the proposed method can improve over 9% of sensitivity, and 14% specificity on the independent dataset in identifying metal-binding cysteines. The proposed method can also improve almost 76% sensitivity with same specificity in metal-binding histidine, and MCC is also improved from 0.28 to 0.88. Our proposed approach achieved a significant improvement with independent test set of metal-binding electron transport proteins. The result shows that our method could be effectively used for indentifying metal-binding sites in electron transport proteins to help biologists better understand the workings of the electron transport chain.
Since user-friendly and publicly accessible web-servers represent the future direction for developing practically more useful models, simulated methods, or predictors, we will make efforts in our future work to provide a web-server for the method presented in this paper.
Funding Statement
This work was partially supported by National Science Council (NSC) of Taiwan, NSC 101-2221-E-155-068 to Yu-Yen Ou. No additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Berg J, Tymoczko JL, Stryer L, Clarke N (2002) Biochemistry. 5th edition. New York: WH Freeman.
- 2. Lin C, Lin K, Yang C, Chung I, Huang C, et al. (2005) Protein metal binding residue prediction based on neural networks. International Journal of Neural Systems 15: 71. [DOI] [PubMed] [Google Scholar]
- 3.Menchetti S, Passerini A, Frasconi P, Andreini C, Rosato A (2006) Improving prediction of zinc binding sites by modeling the linkage between residues close in sequence. Springer LNCS. 309–320.
- 4. Passerini A, Punta M, Ceroni A, Rost B, Frasconi P (2006) Identifying cysteines and histidines in transition metal binding sites using support vector machines and neural networks. Proteins: Structure, Function, and Bioinformatics 65: 305–316. [DOI] [PubMed] [Google Scholar]
- 5. Lippi M, Passerini A, Punta M, Rost B, Frasconi P (2008) MetalDetector: a web server for predicting metal-binding sites and disulfide bridges in proteins from sequence. Bioinformatics 24: 2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chou KC (2011) Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). Journal of Theoretical Biology 273: 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. UniProt Consortium (2010) The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 38: D142–D148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chothia C, Lesk AM (1986) The relation between the divergence of sequence and structure in proteins. The EMBO journal 5: 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu ZC, Xiao X, Chou KC (2012) iLoc-Gpos: A Multi-Layer Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Gram-Positive Bacterial Proteins. Protein and Peptide Letters 19: 4–14. [DOI] [PubMed] [Google Scholar]
- 11. Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292: 195–202. [DOI] [PubMed] [Google Scholar]
- 12. Xie D, Li A, Wang MH, Fan ZW, Feng HQ (2005) LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res 33: W105–W110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ou YY, Gromiha MM, Chen SA, Suwa M (2008) TMBETADISC-RBF: Discrimination of beta-barrel membrane proteins using RBF networks and PSSM profiles. Computational Biology and Chemistry 32: 227–231. [DOI] [PubMed] [Google Scholar]
- 14. Chen SA, Ou YY, Lee TY, Gromiha MM (2011) Prediction of transporter targets using efficient RBF networks with PSSM profiles and biochemical properties. Bioinformatics 27: 2062–2067. [DOI] [PubMed] [Google Scholar]
- 15. Ou YY, Chen SA, Gromiha MM (2010) Prediction of Membrane Spanning Segments and Topology in beta-Barrel Membrane Proteins at Better Accuracy. Journal of Computational Chemistry 31: 217–223. [DOI] [PubMed] [Google Scholar]
- 16. Ou YY, Chen SA, Gromiha MM (2010) Classification of transporters using efficient radial basis function networks with position-specific scoring matrices and biochemical properties. Proteins: Structure, Function, and Bioinformatics 78: 1789–1797. [DOI] [PubMed] [Google Scholar]
- 17. Chou K, Wu Z, Xiao X (2011) iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS ONE 6: e18258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chou KC, Wu ZC, Xiao X (2012) iLoc-Hum: using the accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol BioSyst 8: 629–641. [DOI] [PubMed] [Google Scholar]
- 19. Hayat M, Khan A (2012) MemHyb: Predicting membrane protein types by hybridizing SAAC and PSSM into the general form of Chou's PseAAC. Journal of Theoretical Biology 292: 93–102. [DOI] [PubMed] [Google Scholar]
- 20. Li D, Jiang Z, Yu W, Du L (2010) Predicting Caspase Substrate Cleavage Sites Based on a Hybrid SVMPSSM Method. Protein and Peptide Letters 17: 1566–1571. [DOI] [PubMed] [Google Scholar]
- 21. Chou KC (2004) Structural bioinformatics and its impact to biomedical science. Current Medicinal Chemistry 11: 2105–2134. [DOI] [PubMed] [Google Scholar]
- 22. Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences 89: 10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwartz R, Dayhoff M (1978) Matrices for detecting distant relationships. Atlas of protein sequence and structure 5: 353–358. [Google Scholar]
- 24.Chen SA, Lee TY, Ou YY (2010) Incorporating significant amino acid pairs to identify O-linked glycosylation sites on transmembrane proteins and non-transmembrane proteins. BMC Bioinformatics 11. [DOI] [PMC free article] [PubMed]
- 25.Ou YY (2005) QuickRBF: a package for efficient radial basis function networks. Available: http://csieorg/~yien/quickrbf/. Accessed 2012 Dec 30.
- 26. Ou YY, Oyang YC, Chen CY (2005) A novel radial basis function network classifier with centers set by hierarchical clustering. Proc IJCNN 2005: 1383–1388. [Google Scholar]
- 27. Yang ZR, Thomson R (2005) Bio-basis function neural network for prediction of protease cleavage sites in proteins. IEEE Transactions on Neural Networks 16: 263–274. [DOI] [PubMed] [Google Scholar]
- 28. Zhang GZ, Huang DS (2004) Prediction of inter-residue contacts map based on genetic algorithm optimized radial basis function neural network and binary input encoding scheme. Journal of computer-aided molecular design 18: 797–810. [DOI] [PubMed] [Google Scholar]
- 29.Su CT, Chen CY, Ou YY (2006) Protein disorder prediction by condensed PSSM considering propensity for order or disorder. BMC Bioinformatics 7. [DOI] [PMC free article] [PubMed]
- 30. Lee TY, Chen SA, Hung HY, Ou YY (2011) Incorporating Distant Sequence Features and Radial Basis Function Networks to Identify Ubiquitin Conjugation Sites. PLoS ONE 6: e17331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schneider TD, Stephens RM (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]