

Abstract
The role of microglia in central nervous system pathology has been studied extensively, and more recently, examination of microglia in the healthy brain has yielded important insights into their many functions. It was long assumed that microglia were essentially “quiescent” cells, unless provoked into activation, which was considered a hallmark of disease. More recently, however, it has become increasingly clear that they are extraordinarily dynamic cells, constantly sampling their environment and adjusting to exquisitely delicate stimuli. Along these lines, our lab has identified a new and unexpected role for microglial phagocytosis—or lack thereof—in the pathophysiology of Rett syndrome, a neurodevelopmental disease caused by mutation of the gene encoding methyl-CpG binding protein (MECP)2. Namely, we showed that specific expression of wild-type Mecp2 in myeloid cells of Mecp2-null mice is sufficient to arrest major symptoms associated with this devastating disease. This beneficial effect, however, is abolished if phagocytic activity of microglia is inhibited. Here, we will discuss microglial origins, the role of microglia in brain development and maintenance, and the phenomenon of microglial augmentation by myeloid progenitor cells in the adult brain. Finally, we will address in some detail the beneficial roles of microglia as clinical targets in Rett syndrome and other neurological disorders.
Origins of microglia
The story of microglia is an old one, and as is the case with much of modern neuroscience, Ramon y Cajal is involved in the telling of it. Cajal’s prize student, Pio del Rio-Hortega, is considered the “father” of microglia biology, and his classic work [1] is seminal to the modern understanding of this enigmatic cell type. Microglia are the unique immune-derived denizens of the central nervous system (CNS) parenchyma: neurons, astrocytes, and oligodendrocytes all emanate from neural ectoderm, whereas microglia do not. Rather, it was shown that the microglial precursors, primitive myeloid progenitors, originate outside the CNS in the embryonic yolk sac, and migrate into the brain rudiment at around day 8 during embryogenesis, where they then proliferate until approximately two weeks post-natal [2]. Lineage tracing experiments have further demonstrated that exquisite timing of the expression of runt-related transcription factor (Runx)1 is crucial for the formation of the nascent microglia, and the subsequent establishment of the brain vasculature is necessary for the infiltration of the CNS by these cells [3, 4].
The myeloid origin of microglia and penetration of the brain rudiment to become an intrinsic part of the adult CNS are of considerable interest in terms of microglia function in health, disease, and amelioration of disease. In healthy brain function, their immune ontogeny supports the likelihood that they are ardent phagocytes and cytokine producers. The fact that their ultimate resting place is juxtaneural suggests that they likely play key roles in neuronal maintenance and support; in the case of pathology, however, this intimate relationship with delicate neural cells allows microglia the ability to damage them via production of cytotoxic mediators [5–8]. Their infiltrative and expansive nature during embryogenesis indicates that they are motile cells, apt to respond to molecular gradients and proliferative signals. This infiltrative ability in particular is tantalizing in terms of therapeutic possibilities involving the augmentation of existing microglia by transplanted cells. Although recent work shows clearly that microglial turnover is a case of self-sustaining proliferation rather than supplementation by circulating monocytes [3, 9], it has also been shown that in the special case of brain irradiation followed by bone marrow transplant that myeloid progenitor cells are able to engraft within the brain parenchyma and assume functions remarkably similar to those performed by true microglia [10, 11]. This phenomenon suggests the possibility of harnessing immune-based strategies as a therapeutic modality to deliver cellular or molecular therapies in CNS pathologies.
Microglia: role in the developing and adult brain
Long thought to be inactive except in response to pathological insults such as injury or infection, microglia have since been demonstrated to be dynamic immune effector cells [12, 13] even in their normal quiescent or so-called “resting” state in the brain. Microglia, like peripheral myeloid-derived cells, phagocytose debris and secrete cytokines and chemokines [14], the signature chemical messengers of immune cells. Recent work shows them to be intimately involved in the sculpting of the CNS.
Phagocytosis of apoptotic cells is evolutionarily conserved from the nematode to mammals [15–17], and is a necessary part of normal tissue development [18]. Microglial removal of supernumerary Purkinje neurons was demonstrated in early postnatal cerebellar slice cultures wherein microglia contacted and engulfed the majority of cleaved caspase-3 expressing cells [19]. Deficiencies in microglial activation proteins CD11b and DAP12 were correlated with impairment in developmental apoptosis in murine hippocampus [20]. Importantly, this coupling of neuronal apoptosis and microglial phagocytosis is not limited to early development; microglia were demonstrated to be the primary phagocytes of apoptotic neural progenitor cells in the adolescent sub-granular zone of the hippocampus, a primary neurogenic niche [21]. Interestingly, this effect was linked to microglia that were fully ramified, thus exhibiting the morphological signature of a resting cell within the normal CNS milieu, rather than the more amoeboid morphology of a cell responding to “threat.”
In addition to removing whole cells from the CNS, microglia also participate in synapse removal during development. Microglia were shown by stimulated emission depletion (STED) and electron microscopy to contain markers unique to the neuronal post-synaptic density [22]. Microglial phagocytosis was subsequently demonstrated in the developing visual system to be activity-dependent, and dynamic partners with neurons in response to sensory stimuli [23]. Recognition and engulfment of pre-synaptic material was suggested to be reliant upon the recognition of immune complement factors, including C1q and C3, by receptors unique in the CNS parenchyma to microglia [24, 25]. Importantly, these phenomena, when disrupted, were associated with significant changes in synaptic connectivity—strongly suggesting their physiologic importance in circuit building and plasticity.
Microglia have been demonstrated to be players in connectivity by virtue of their production of cytokines, a primary function of immune cells. Microglia are a significant source of cytokine production within the CNS milieu, crucial for neural/glial specification. Several immune cytokines produced by microglia, including (but not limited to) interleukin (IL)-1β, tumor necrosis factor (TNF), and IL-6 are expressed in the developing CNS [26]. IL-6, for example, signals through the glycoprotein (gp) 130 receptor, and affects transcription of genes responsible for determining the fate of neuroepithelial cells as radial glial cells, neurons, or glia [27]. Mice deficient in TNF were shown to exhibit marked reduction in neuronal arborization in both CA1 and CA3 regions of hippocampus, and significant impairment in hippocampal-dependent behavioral assays [28]. The more recent finding that activated microglia facilitate the induction of long-term potentiation (LTP—a basic underpinning of neuronal memory formation) in CNS by release of TNF is in agreement with this notion of TNF as a microglial-derived synaptic modulator [29].
Thus, immune dysfunction perturbing normal phagocytic capability by microglia during pre- or post-natal CNS development is likely to have serious ramifications in terms of emergent CNS function [18] A dysregulated immune response may contribute to pathologies seen in neurodevelopmental disorders involving aberrant dendritic arborization and spines.
Microglia in tissue homeostasis, repair and protection
Tissue-resident cells of the monocytic lineage, Kupffer cells for example, contribute to the critical jobs of tissue repair, remodeling, growth, and homeostasis through the secretion of anti-inflammatory cytokines, growth factors, and by clearance of debris that would otherwise become toxic to surrounding tissue [30]. In turn, pro-inflammatory mediators can alter the phenotype of any mononuclear phagocyte, including microglia, to become inflammatory. Overall, the balance between pro- and anti-inflammatory phenotypes is essential for longevity and health.
Classically activated macrophages secrete pro-inflammatory cytokines and release nitric oxide following expression of inducible nitric oxide synthase (iNOS) [31, 32]. As can be presumed from their cellular products, this subset is induced in response to acute insult. Subsequently, alternatively-activated macrophages are essential for proper resolution of inflammation and tissue repair [32–34].
While classical vs. alternative activation of peripheral macrophages is well-studied, similar paradigms have not yet been widely established in microglia. Therefore, whether microglial skewing phenotypes mirror those of macrophages, or if microglia have a unique set of activation paradigms in vivo, is relatively unknown and cannot be assumed. It has been suggested that microglia and peripheral macrophages respond to polarizing stimuli similarly [35]. Thus we hypothesize that microglial phenotypes in vivo may reflect varying degrees of pro-inflammatory, anti-inflammatory, tissue-remodeling and growth-promoting behaviors. Still, microglia are not peripheral macrophages, and the CNS represents a unique immunologic environment. The scope of this opinion and the relative paucity of knowledge regarding microglial skewing preclude us from speculating on these phenotypes in further detail, but there is an overall agreement in the field that microglia are involved with tissue maintenance and homeostasis and can assume pro-inflammatory activation in response to threat (Figure 1).
Figure 1.
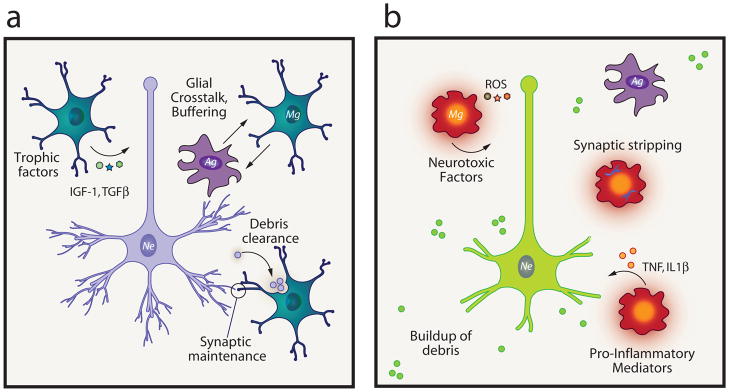
(a) Unchallenged microglia perform functions in the healthy CNS including phagocytosis of normal apoptotic debris, secretion of trophic factors, including IGF-1 and TGFβ, synaptic maintenance, and roles in CNS development (not shown). (b) Challenged microglia respond to CNS damage, either sterile (injury, stroke) or pathogen-mediated by secreting pro-inflammatory factors, including TNFα and IL-1β and reactive oxygen species (ROS). When these mediators persist unchecked, neurodegeneration can ensue. Synaptic stripping is seen immediately following inflammation.
Mg=microglia, Ag=astroglia, Ne=neuron, ROS=reactive oxygen species, IGF-1=insulin-like growth factor 1, TGFβ=transforming growth factor beta, TNF=tumor necrosis factor, IL1β=interleukin 1 beta.
Uptake of apoptotic cells by professional phagocytes leads to downregulation of pro-inflammatory mediators such as IL-1β and TNF, and increased production of anti-inflammatory cytokines such as transforming growth factor beta (TGFβ), driving local immunosuppression [36]. This anti-inflammatory response to phagocytosis of apoptotic cells has also been shown in microglia [37]. This mechanism, which attenuates the immune response subsequent to damage clearance of “self,” prevents immune-mediated tissue destruction. It should be noted, however, that microglia stimulated with molecules such as lipopolysaccharide (LPS) or β-amyloid induce neuronal death by phagocytosis of otherwise viable neurons in vitro [38]. It was also shown both in vitro and in vivo that milk fat globule-EGF factor 8 protein (MFG-E8), a bridging molecule between phosphatidylserine exposed on apoptotic cells and the vitronectin receptor on phagocytes, mediates phagocytosis and the resulting death of otherwise viable neurons in an LPS-induced model of neuroinflammation [39]. This suggests that the presence of a pro-inflammatory mediator may alter the behavior of microglia from a quiescent to an inflammatory, potentially destructive phenotype. They also illustrate that phagocytosis can be a destructive rather than protective process in the CNS [39–42]. Microglial impact on surrounding neurons may also be a product of non-immune cell initiated action. For example, it was shown that following ablation of sensory neurons in olfactory bulb, activated microglia were primarily responsible for spontaneous clearance of neurons deprived of sensory input [43]. Such mutifactorial interactions complicate our understanding of microglial function in health and disease [44].
Activated microglia were primarily assumed to be neurodestructive [45], and ample evidence supports this contention. Works in diverse models feature microglia as endowed with destructive potential [46–48]. However, recent studies have begun to elucidate specific molecular players involved in the delicate balance between damaging and supportive microglial phenotypes, and suggest a more complex picture. For example, it was shown in models of Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS), as well as subsequently in spinal cord injury, that the fractalkine receptor, CX3CR1, was a key mediator of inflammation and subsequent neurodegeneration [49]; similarly, chemokine receptor type 2 (CCR2) deficiency accompanied impaired microglial accumulation and increased severity of pathology in a model of Alzheimer’s disease (AD) [50].
Along these lines, recent works implicate microglia [51, 52], as well as blood-borne myeloid cells [33] in neuroprotective roles. Microglia were shown to engage with neurons after ischemic insult and provide protection against oxygen and glucose deprivation, possibly by an integrin-dependent mechanism [53]. Acute ablation of microglia in organotypic hippocampal slice cultures was associated with significant exacerbation of neuron death after excitotoxic challenge [54].
In terms of possible peripheral immune influence on microglial activity, it may be important that microglia comprise a small but significant percentage [55] of the glia limitans—a structure which is proximal to both brain and circulation—and respond robustly to cytokine stimulation. Thus, they are in a position to be affected by peripherally secreted mediators and possibly transduce signals affecting neural cells within the brain itself. For example, microglia stimulated by IL-4—a cytokine produced predominantly by circulating immune cells—produce insulin-like growth factor (IGF)-1 [56], in support of neural growth and cognitive function. IGF-1 has been shown to be neuroprotective in several disease models including those for stroke [57], ALS [58], PD [59], and multiple sclerosis (MS). Using the experimental autoimmune encephalitis (EAE) model of MS, the striking observation was made that a specific microRNA, miR-124, was oppositely expressed in macrophages and microglia and was also a key regulator of supportive vs. destructive phenotype of these cells [60].
In sum, these works suggest that the function of microglia is exceedingly complex, and that depending upon milieu and molecular mediators, they can be devastatingly destructive, or surprisingly ameliorative in pathological situations.
Microglia as therapeutic targets and effectors
Because microglia play key roles in CNS development, tissue homeostasis, synaptic regulation and neural plasticity, these cells may be attractive therapeutic targets either directly, by pharmaceutical strategies, or by augmentation via bone marrow transplantation. Indeed, it has been shown by several groups that, following conditioning by brain irradiation, hematopoietic precursor cells can migrate to the brain and assume roles and morphology similar to resident microglia [61–63]. Prinz and coworkers demonstrated that the precursors for these “microglia-like” cells were predominantly Ly6chiCCR2+ in phenotype, and that irradiation was a key factor to induce their engraftment [11]. Accordingly, in a model radically different from bone marrow transplantation (i.e. parabiosis—in which donor and recipient mice are connected via the circulatory system), Rossi and coworkers showed that in situ proliferation of existing local microglial cells was the driver of turnover, and that in the absence of bone marrow transplantation with irradiation, circulating monocyte precursors did not contribute significantly to adult microgliogenesis [9].
The approach of utilizing bone marrow transplant to address CNS pathology was first used in the twitcher mouse, a model for globoid cell leukodystrophy [64]—an often-fatal lipid storage disorder featuring severe neurological pathology [65]. It was shown that following bone marrow transplantation, engraftment of peripheral organs and CNS by donor-derived cells accompanied CNS remyelination in treated mice [66]. In the years since, the approach has been translated from bench to bedside with a reasonable degree of success: cord blood transfusions into infants, or bone marrow transplantation into presymptomatic cases and mild later-onset cases have demonstrated significant improvement in human patients [67]. Similar strategies have been used with other metabolic storage diseases with varying efficacy [68].
Microglia in Rett syndrome
Rett syndrome is an X-linked CNS developmental disorder primarily affecting girls. In the majority of cases, Rett syndrome is linked to mutations in the gene MECP2 [69], which acts to modulate transcription of many other genes [70]. Patients with Rett display psychomotor impairment, motor deficits and stereotyped behaviors, tremors, apneas, and impaired language development [71]. Neurons in Rett patients exhibit decreased dendritic arborization, fewer dendritic spines, and increased cell packing density [72, 73]. MECP2, in addition to its role in the etiology of Rett syndrome, has been suggested to play a simultaneous role in the immune system regulation [74–76]. Nevertheless, research into immune system abnormalities in Rett has been limited.
Whereas initial studies strongly supported an exclusively neuronal role for Mecp2 in Rett pathology, it has since been shown that other cells, specifically glia, play a major role in the disease [77, 78]. Genetic manipulations in mouse models were used to demonstrate that specific neuron-restricted expression of wild-type Mecp2 protein is insufficient for complete rescue of all disease phenotype [77, 79]; ubiquitous expression is necessary [80]. Later studies disclosed that Mecp2 is also expressed in glia, and that expression of wild-type Mecp2 in astrocytes was subsequently shown to arrest disease progression in otherwise Mecp2-null mice [81], thus opening to question intrinsic neuronal dysfunction as the sole mediator of this disease. Around the same time, Mecp2-null microglia were suggested to be toxic to neurons by virtue of upregulated secretion of glutamate [82].
In parallel, our group had also been conducting studies to address the role of microglial immunity in Rett syndrome. We had selected a strategy of bone marrow transplant in four-week old Mecp2-null male mice. This time-point was chosen to precede onset of severe symptoms, but to be long enough post-natal to avoid major neurodevelopmental milestones. By 8–10 weeks post bone marrow transplantation, robust engraftment of microglia-like cells from donor mice was evident in brains of Mecp2-null animals. To our surprise, pathology was halted; mice with an expected lifespan of eight weeks survived to nearly a year. These animals were active, well-groomed, and showed remarkable improvement in several characteristic disease phenotypes including apneas, tremors and locomotor performance. Of critical importance was the observation that Mecp2-null males that received Mecp2-null bone marrow fared as poorly as non-transplanted mice, indicating that myeloablative conditioning of the brain itself was unlikely playing a role in disease amelioration—a possibility given recent findings suggesting direct effects of brain irradiation on microglial morphology and proximity to phagocytic targets in a model of AD [10].
Examining microglia from Mecp2-null mice and wild-type controls in vitro, striking impairment in phagocytic ability of primary Mecp2-null microglia was apparent. Thus, we considered the possibility that insufficient clearance of debris within the brains of these animals might indeed contribute to the severity of pathology. To test this, we drove expression of wild-type Mecp2 in myeloid cells of Mecp2-null mice. Similar to bone marrow transplanted mice, LysmcreMecp2lox-stop/y mice exhibited disease arrest. Most interestingly, chronic injection of annexin V into these animals abolished the benefit conferred by wild-type microglia, and was associated with a buildup of apoptotic CNS debris [83]. These results suggest that phagocytic clearance by microglia was important to amelioration of disease (Figure 2).
Figure 2.

(a) Possible benefits to Mecp2-null neural cells in the presence of normally-functioning wild-type microglia including microglial secretion of trophic factors (IGF-1, BDNF), and apoptotic debris clearance. Microglial support of astrocytes may also be important since normally-functioning astrocytes have been shown to ameliorate Rett pathology in a similar fashion (See Ref. 81). In the presence of annexin V (denoted as “a” in figure), phosphatidylserine residues on apoptotic debris are blocked. Over time, debris buildup leads to a suboptimal CNS milieu, and microglia may convert to a neurodestructive phenotype, releasing pro-inflammatory and neurotoxic mediators (see Refs. 77, 82); accordingly, neurons become impaired in function.
KO=Mecp2-null, Mg=microglia, Ag=astroglia, Ne=neuron, IGF-1= insulin-like growth factor 1, BDNF=brain-derived neurotrophic factor, TNF=tumor necrosis factor, a=annexin V.
Although data supporting the importance of phosphatidylserine recognition (which would be blocked by annexin administration) in this model is strong, other possible mediators of phagocytosis should also be investigated, including P2Y6 receptor signaling, deficiencies in which might interfere with recognition of the nucleotide UDP released from injured neurons. [84]. In combination with increased levels of neurotoxic glutamate, suggested by Jin and coworkers [77, 82], a scenario in which microglia first induce injury then subsequently fail to properly remove neuronal debris might be reasonably hypothesized. Triggering receptor expressed on myeloid cells (TREM)2 signaling has also been shown to facilitate debris clearance by microglia [85]. Interestingly, TREM2 dysfunction was implicated in Nasu-Hakola disease. Nasu-Hakola features progressive dementia and bone cysts [86], suggesting parallels to the Rett syndrome patients who, in addition to primary neurological sequelae frequently suffer from osteopenia and scoliosis [87, 88]. Thus, similar mechanisms may be relevant to other diseases, and we hypothesize that impaired phagocytic activity of microglia may contribute to pathophysiology of other CNS disorders.
These data have implications for the treatment of Rett syndrome. Pre-clinical work has largely focused in two directions: a gene-therapy approach [89, 90] and a growth factor supplementation approach [91, 92]. The idea of targeting the immune system in general and microglia in particular is new and promising because myeloid cells are relatively straightforward to isolate and manipulate ex vivo, and are able to enter the CNS via innate mechanisms. In clinical practice, a sensible approach might be ex vivo sorting of the patient’s own bone marrow into mutant and normal cells, with reinjection following myeloablative conditioning. Following initial engraftment, a secondary treatment involving “softening” of the blood-brain barrier (BBB) combined with rigorously controlled local CNS inflammation [93] could be advantageous, thus allowing maximal entrance of hematopoietic cells and subsequent engraftment of microglia-like cells. If Rett syndrome is truly both a CNS and an immune system pathology, a treatment that takes into account the entire body, i.e. immune system replacement, would be well-suited as a clinically-feasible and long-term therapy for this devastating disorder.
Concluding remarks
Immune mediators, both central to and peripheral to the CNS, have now been shown to mediate CNS processes as fundamental as neuronal guidance, as subtle as synaptic refinement, and as complex as learning and memory [25, 94, 95]. Even within the realm of disease, the purview of the neuroimmunologist is now considerably extended beyond “core” neuroinflammatory disorders. Pathologies such as PD [96], AD [97], and Huntington’s disease [98] have all been demonstrated to feature a degree of immune dysregulation as contributing factors, and the list is ever-growing. Psychiatric disorders, including major depression, bipolar disorder, and schizophrenia, all of which have previously been attributed to CNS-exclusive dysfunction, are now suggested to have strong immune links [99–101].
Therefore, our understanding of the term “neuroimmunology,” and what it encompasses, has changed remarkably. Once concerned exclusively with a small cohort of pathological conditions involving clear attack of the CNS by the immune system, neuroimmunologists are now beginning to uncover healthy physiological interactions between the two systems that have implications far beyond the setting of pathology [94, 102]. Immune malfunction should be routinely considered as a potentially contributory or causative factor in the etiology of neurodevelopmental, cognitive, and psychiatric dysfunction.
Acknowledgments
Noël C. Derecki is a recipient of a Hartwell Foundation post-doctoral fellowship. This work was primarily supported by the Rett Syndrome Research Trust (RSRT) and by the National Institute of Neurological Disorders and Stroke, NIH (NS081026) to J. K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rio-Hortega D. Microglia. P. B. Hoebaer; New York, N.Y: 1932. Cytology and cellular pathology of the nervous system; pp. 483–534. [Google Scholar]
- 2.Alliot F, et al. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain research Developmental brain research. 1999;117:145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 3.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 5.Cui Y, et al. Neuroprotective effect of methyl lucidone against microglia-mediated neurotoxicity. European journal of pharmacology. 2012;690:4–12. doi: 10.1016/j.ejphar.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 6.Mead EL, et al. Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. Journal of neurochemistry. 2012;121:287–301. doi: 10.1111/j.1471-4159.2012.07659.x. [DOI] [PubMed] [Google Scholar]
- 7.Filipov NM, Dodd CA. Role of glial cells in manganese neurotoxicity. J Appl Toxicol. 2012;32:310–317. doi: 10.1002/jat.1762. [DOI] [PubMed] [Google Scholar]
- 8.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ajami B, et al. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 10.Mildner A, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mildner A, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 12.Nimmerjahn A, et al. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 13.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature neuroscience. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 14.Aloisi F, et al. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte-macrophage colony-stimulating factor and interaction with Th1 cells. J Immunol. 2000;164:1705–1712. doi: 10.4049/jimmunol.164.4.1705. [DOI] [PubMed] [Google Scholar]
- 15.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010;464:778–782. doi: 10.1038/nature08853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinchen JM, Ravichandran KS. Phagocytic signaling: you can touch, but you can’t eat. Curr Biol. 2008;18:R521–524. doi: 10.1016/j.cub.2008.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roth KA, D’Sa C. Apoptosis and brain development. Ment Retard Dev Disabil Res Rev. 2001;7:261–266. doi: 10.1002/mrdd.1036. [DOI] [PubMed] [Google Scholar]
- 19.Marin-Teva JL, et al. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- 20.Wakselman S, et al. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sierra A, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 23.Tremblay ME, et al. Microglial interactions with synapses are modulated by visual experience. PLoS biology. 2010;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 26.Mehler MF, Kessler JA. Hematolymphopoietic and inflammatory cytokines in neural development. Trends in Neurosciences. 1997;20:357–365. doi: 10.1016/s0166-2236(96)01045-4. [DOI] [PubMed] [Google Scholar]
- 27.Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Golan H, et al. Involvement of tumor necrosis factor alpha in hippocampal development and function. Cerebral cortex. 2004;14:97–105. doi: 10.1093/cercor/bhg108. [DOI] [PubMed] [Google Scholar]
- 29.Zhong Y, et al. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-alpha. Brain, behavior, and immunity. 2010;24:874–880. doi: 10.1016/j.bbi.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards JP, et al. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Shechter R, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz M. “Tissue-repairing” blood-derived macrophages are essential for healing of the injured spinal cord: from skin-activated macrophages to infiltrating blood-derived cells? Brain Behav Immun. 2010;24:1054–1057. doi: 10.1016/j.bbi.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Michelucci A, et al. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Fadok VA, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magnus T, et al. Microglial phagocytosis of apoptotic inflammatory T cells leads to down-regulation of microglial immune activation. J Immunol. 2001;167:5004–5010. doi: 10.4049/jimmunol.167.9.5004. [DOI] [PubMed] [Google Scholar]
- 38.Neher JJ, et al. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973–4983. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- 39.Fricker M, et al. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:2657–2666. doi: 10.1523/JNEUROSCI.4837-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neher JJ, et al. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. Journal of immunology. 2011;186:4973–4983. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- 41.Fricker M, et al. Primary phagocytosis of viable neurons by microglia activated with LPS or Abeta is dependent on calreticulin/LRP phagocytic signalling. Journal of neuroinflammation. 2012;9:196. doi: 10.1186/1742-2094-9-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bessis A, et al. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- 43.Lazarini F, Lledo PM. Is adult neurogenesis essential for olfaction? Trends in Neurosciences. 2011;34:20–30. doi: 10.1016/j.tins.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Glass CK, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Block ML, et al. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 46.Wu DC, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Aversa TG, et al. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. Journal of neurovirology. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- 48.Block ML, et al. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 49.Cardona AE, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nature neuroscience. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 50.El Khoury J, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature medicine. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 51.Butovsky O, et al. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Persson M, et al. Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-alpha. Glia. 2005;51:111–120. doi: 10.1002/glia.20191. [DOI] [PubMed] [Google Scholar]
- 53.Neumann J, et al. Microglia provide neuroprotection after ischemia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- 54.Vinet J, et al. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. Journal of neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lassmann H, et al. Microglial cells are a component of the perivascular glia limitans. J Neurosci Res. 1991;28:236–243. doi: 10.1002/jnr.490280211. [DOI] [PubMed] [Google Scholar]
- 56.Butovsky O, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2005 doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 57.Dluzniewska J, et al. A strong neuroprotective effect of the autonomous C-terminal peptide of IGF-1 Ec (MGF) in brain ischemia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1896–1898. doi: 10.1096/fj.05-3786fje. [DOI] [PubMed] [Google Scholar]
- 58.Kaspar BK, et al. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 59.Ebert AD, et al. Human neural progenitor cells over-expressing IGF-1 protect dopamine neurons and restore function in a rat model of Parkinson’s disease. Experimental neurology. 2008;209:213–223. doi: 10.1016/j.expneurol.2007.09.022. [DOI] [PubMed] [Google Scholar]
- 60.Ponomarev ED, et al. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nature medicine. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ladeby R, et al. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 62.Flugel A, et al. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity. 2001;14:547–560. doi: 10.1016/s1074-7613(01)00143-1. [DOI] [PubMed] [Google Scholar]
- 63.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 64.Kobayashi T, et al. The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease) Brain research. 1980;202:479–483. doi: 10.1016/0006-8993(80)90159-6. [DOI] [PubMed] [Google Scholar]
- 65.Kohlschutter A, Eichler F. Childhood leukodystrophies: a clinical perspective. Expert review of neurotherapeutics. 2011;11:1485–1496. doi: 10.1586/ern.11.135. [DOI] [PubMed] [Google Scholar]
- 66.Hoogerbrugge PM, et al. Donor-derived cells in the central nervous system of twitcher mice after bone marrow transplantation. Science. 1988;239:1035–1038. doi: 10.1126/science.3278379. [DOI] [PubMed] [Google Scholar]
- 67.Suzuki K. Globoid cell leukodystrophy (Krabbe’s disease): update. Journal of child neurology. 2003;18:595–603. doi: 10.1177/08830738030180090201. [DOI] [PubMed] [Google Scholar]
- 68.Krivit W. Stem cell bone marrow transplantation in patients with metabolic storage diseases. Advances in pediatrics. 2002;49:359–378. [PubMed] [Google Scholar]
- 69.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 70.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shetty AK, et al. Syndrome of microcephaly, mental retardation, and tracheoesophageal fistula associated with features of Rett syndrome. J Child Neurol. 2000;15:61–63. doi: 10.1177/088307380001500114. [DOI] [PubMed] [Google Scholar]
- 72.Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- 73.Noutel J, et al. Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron. 2011;70:35–42. doi: 10.1016/j.neuron.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tong Y, et al. T-bet antagonizes mSin3a recruitment and transactivates a fully methylated IFN-gamma promoter via a conserved T-box half-site. Proc Natl Acad Sci U S A. 2005;102:2034–2039. doi: 10.1073/pnas.0409510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lal G, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balmer D, et al. MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum Genet. 2002;110:545–552. doi: 10.1007/s00439-002-0724-4. [DOI] [PubMed] [Google Scholar]
- 77.Maezawa I, et al. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ballas N, et al. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alvarez-Saavedra M, et al. Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. Hum Mol Genet. 2007;16:2315–2325. doi: 10.1093/hmg/ddm185. [DOI] [PubMed] [Google Scholar]
- 80.Guy J, et al. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lioy DT, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Derecki NC, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koizumi S, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takahashi K, et al. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. The Journal of experimental medicine. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paloneva J, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. American journal of human genetics. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hofstaetter JG, et al. Altered bone matrix mineralization in a patient with Rett syndrome. Bone. 2010;47:701–705. doi: 10.1016/j.bone.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 88.Han ZA, et al. Clinical characteristics of children with rett syndrome. Ann Rehabil Med. 2012;36:334–339. doi: 10.5535/arm.2012.36.3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rastegar M, et al. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS ONE. 2009;4:e6810. doi: 10.1371/journal.pone.0006810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jugloff DG, et al. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Human molecular genetics. 2008;17:1386–1396. doi: 10.1093/hmg/ddn026. [DOI] [PubMed] [Google Scholar]
- 91.Tropea D, et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deogracias R, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laflamme N, et al. Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J Neurochem. 2001;79:648–657. doi: 10.1046/j.1471-4159.2001.00603.x. [DOI] [PubMed] [Google Scholar]
- 94.Derecki NC, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–1080. doi: 10.1084/jem.20091419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huh GS, et al. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hamza TH, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oddo S, et al. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 98.Kwan W, et al. Bone marrow transplantation confers modest benefits in mouse models of Huntington’s disease. J Neurosci. 2012;32:133–142. doi: 10.1523/JNEUROSCI.4846-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dantzer R, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kipnis J, et al. Loss of autoimmune T cells correlates with brain diseases: possible implications for schizophrenia? Trends Mol Med. 2006;12:107–112. doi: 10.1016/j.molmed.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 101.Kaminsky Z, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.64. [DOI] [PubMed] [Google Scholar]
- 102.Ziv Y, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]