

Abstract
The prevalence of the metabolic syndrome and nonalcoholic fatty liver disease (NAFLD) in humans increases with age. It is unknown whether this association is secondary to the increased incidence of risk factors for NAFLD that occurs with aging, reflects the culmination of years of exposure to lifestyle factors such as a high-fat diet (HFD), or results from physiological changes that characterize aging. To examine this question, the development of NAFLD in response to a fixed period of HFD feeding was examined in mice of different ages. Mice aged 2, 8 and 18 months were fed 16 weeks of a low-fat diet or HFD. Increased body mass and insulin insensitivity occurred in response to HFD feeding irrespective of the age of the mice. The amount of HFD-induced hepatic steatosis as determined biochemically and histologically was also equivalent among the three ages. Liver injury occurred exclusively in the two older ages as reflected by increased serum alanine aminotransferase levels, positive TUNEL staining and caspase activation. Older mice also had an elevated innate immune response with a more pronounced polarization of liver and adipose tissue macrophages into an M1 phenotype. Studies of cultured hepatocytes from young and old mice revealed that aged cells were selectively sensitized to the Fas death pathway.
Conclusion
Aging does not promote the development of hepatic steatosis but leads to increased hepatocellular injury and inflammation that may be due in part to sensitization to the Fas death pathway and increased M1 macrophage polarization.
Keywords: Fas, liver injury, macrophages, metabolic syndrome, nonalcoholic fatty liver disease
Nonalcoholic fatty liver disease (NAFLD) represents a continuum from simple hepatic steatosis alone to steatosis together with hepatitis and eventual fibrosis.1 The mechanisms underlying the development of this disease remain unclear, but NAFLD is considered the hepatic manifestation of the metabolic syndrome and therefore linked to the development of obesity and insulin resistance.2 The prevalence of the metabolic syndrome increases markedly with aging.3 In 20 to 29 year olds the prevalence is 7% but rises to 44% in individuals aged 60 to 69.3 Similarly aging has also been shown to be a risk factor for the development of human NAFLD,4,5 although some studies have suggested that this effect is limited to females.6,7 More conclusive of an effect of aging on NAFLD is evidence that the prevalence of nonalcoholic steatohepatitis (NASH) and chronic liver disease is increased in the aged. Thus, steatohepatitis8 and fibrosis9,10 are increased with aging leading to a higher mortality in aged individuals with NAFLD.11,12
Aging may promote the development of NAFLD through several mechanisms. First, aging promotes the onset of obesity and diabetes and the increase in these known risk factors for NAFLD may account for the higher prevalence of NAFLD in the aged. Supportive of this possibility is that in parallel with steep rises in obesity and insulin resistance in the pediatric population, problems that had formerly been restricted to the elderly, there has been a dramatic increase in the incidence of NAFLD in young individuals. Second, the increase in disease with age may result from the cumulative effects over many years of lifestyle or environmental factors such as the over consumption of a Western diet. Finally, physiological changes inherent to the process of aging may trigger or promote the development of components of the metabolic syndrome later in life. For example, function of the lysosomal degradative pathway of autophagy decreases with aging,13 and adequate levels of hepatic autophagy are critical to limit lipid accumulation in the liver.14,15 Another possibility is that aging is associated with increased oxidative stress,16 which has also been implicated as a mediator of NAFLD.17 Several of the known, or even currently unknown, physiological changes that occur with aging may therefore lead to NAFLD.
To begin to examine the potential effects of aging on NAFLD development, we challenged mice of different ages with a defined period of high-fat diet (HFD) feeding to induce the metabolic syndrome and NAFLD. This design was intended to ask whether the aging state altered the development of steatosis and hepatitis in response to a defined period of dietary factors that induce NAFLD. Age failed to affect the development of metabolic abnormalities such as weight gain and insulin resistance. The amount of steatosis was similarly unaffected by age. However, aged mice developed more liver injury and a greater inflammatory response from a HFD. Studies in cultured hepatocytes revealed that cells from aged mice were sensitized to death from stimulation of the Fas death pathway. Aging therefore does not affect the development of simple steatosis but promotes the progression to steatohepatitis.
Materials and Methods
Animal Model
Male C57BL/6 mice aged 2, 8 and 18 months were obtained from the aging colony maintained by the National Institute of Aging (Bethesda, MD). Mice were maintained under 12 h light/dark cycles with unlimited access to food and water. Mice were fed either a low-fat diet (LFD) (10% kcal supplied by fat; Research Diets #D12450B; New Brunswick, NJ) or a HFD (60% kcal supplied by fat; Research Diets #D12492) for 16 weeks prior to sacrifice. All studies were approved by the Animal Care and Use Committee of the Albert Einstein College of Medicine and followed the National Institutes of Health guidelines for animal care.
Histology
Livers were fixed in 10% neutral formalin and 6 μm sections stained with hematoxylin and eosin. Tissue sections were examined in a blinded fashion by a single pathologist and graded for the degree of hepatic steatosis. Hepatocellular steatosis was semiquantitatively graded on a sliding scale of: 0, absent; 0.5, minimal; 1, mild; 1.5, mild to moderate; 2, moderate; 2.5, moderate to marked; and 3, marked.
Serum Assays
Serum glucoses were assayed with an Ascensia Contour glucose meter (Bayer HealthCare, Mishawaka, IN). Serum insulin levels were measured by radioimmunoassay, as previously described.18 Relative insulin sensitivity was determined by the homeostasis model assessment of insulin resistance (HOMA), as previously described.19 Alanine aminotransferase (ALT) levels were measured by commercial kit (TECO Diagnostics, Anaheim, CA).
Triglyceride (TG) Assay
Hepatic TG content was determined by commercial kit according to the manufacturer’s instructions (Sigma, St. Louis, MO). TG values were normalized to liver weight.
Terminal Deoxynucleotide Transferase-mediated Deoxyuridine Triphosphate Nick End-labeling (TUNEL) Assay
The numbers of TUNEL positive cells were determined in liver sections with the DeadEnd Colorimetric System (Promega, Madison, WI). Tissue sections were deparaffinized in xylene, rehydrated in decreasing concentrations of ethanol, and the assay performed according to the manufacturer’s instructions. Under light microscopy, the number of TUNEL positive cells in 20 randomly selected high power fields was determined as a percentage of the total number of hepatocytes.
Real-time Reverse Transcription Polymerase Chain Reaction
RNA was isolated from livers or adipose tissue with RNeasy Plus (QIAGEN, Valencia, CA). Reverse transcription was carried out with 1 μg of RNA in an Eppendorf Mastercycler (Hamburg, Germany) using a high capacity cDNA reverse transcription kit (ABI, Foster City, CA). Annealing of primers was done at 25°C for 10 min, followed by elongation at 37°C for 2 h and inactivation of the enzyme at 85°C for 5 min. Negative controls (no added transcriptase) were performed in parallel. PCRs for tumor necrosis factor-α (TNF), monocyte chemoattractant protein 1 (MCP-1), F4/80, nitric oxide synthase 2 (NOS2), arginase 1, Fas, FasL and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were performed in triplicate in a 7500 Fast Real-Time PCR System (ABI). Primers to these genes (Supplemental Table 1) were purchased from Integrated DNA Technologies (Coralville, IA). PCR was carried out using Power SYBR Green Master Mix (ABI). Taq polymerase was activated at 95°C for 10 min. The cycling parameters were denaturation at 95°C for 30 sec and extension at 60°C for 1 min (for 40 cycles). Data analysis was performed using the 2−ΔΔCT method for relative quantification. All samples were normalized to GAPDH which served as an endogenous control.
Primary Hepatocyte Isolation and Culture
Primary hepatocytes from C57BL/6 mice of 2 or 18 months of age were obtained by collagenase perfusion, as previously described.20 The cells were purified by Percoll (Sigma) density centrifugation and cultured in RPMI 1640 with 5% fetal bovine serum, 1.7 μM insulin, 1 μM dexamethasone and antibiotics on Biocoat plates (Becton Dickinson, Franklin Lakes, NJ). The medium was changed 3 h after plating and again at 18 h at which time the experiments were started. Some of the cells were treated with: 40–60 μM menadione (Sigma); 15 ng/ml TNF (R&D Systems, Minneapolis, MN) alone or after a 1 h pretreatment with 50 nM actinomycin D (Sigma); 0.25 or 0.5 mM palmitate conjugated with bovine serum albumin (Sigma), as previously described;21 or 25 or 100 ng/ml of Jo2 antibody (BD Pharminogen, San Diego, CA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
The percentage cell death in cultured hepatocytes was quantified by MTT assay.22 At 24 h after treatment with a death stimulus, the cell culture medium was replaced by an equal volume of a 1 mg/ml MTT solution, pH 7.4, in RPMI 1640. After incubation at 37°C for 1 h, the MTT solution was discarded, the formazan product solubilized with 1-propanol and the absorbance of this compound measured at 560 nm in a plate reader. The percentage cell death was determined by dividing the optical density of the treated group by the optical density for untreated, control cells, multiplying by 100, and subtracting that number from 100.
Fluorescence Microscopy
Numbers of apoptotic and necrotic hepatocytes were determined by fluorescence microscopy of acridine orange and ethidium bromide costained cells, as previously described.23 Cells with fragmented or condensed nuclei and a shrunken cytoplasm by acridine orange staining were considered apoptotic. Necrotic cells were detected by positive staining with ethidium bromide. A minimum of 300 cells per sample were examined, and numbers of apoptotic and necrotic cells expressed as a percentage of the total number of cells.
Statistical Analysis
Numerical results are reported as mean ± S.E. and represent at least three independent experiments. Control and experimental groups were compared by the unpaired Student’s t-test with statistical significance defined as P<0.05.
Results
Effects of Aging on Body and Fat Mass and Insulin Sensitivity
C57BL/6 mice were started on either a LFD or HFD at 2, 8 and 18 months of age. These ages were selected to reflect the normal physiology of young, middle-aged and old mice. The mice were sacrificed for analysis after 16 weeks on their respective diets at the ages of 6, 12 and 22 months. Modest weight gain (19–31%) occurred in LFD-fed mice, with the greatest increase occurring in the 6-month old animals (Fig. 1A). The HFD induced a highly significant increase in body mass (71–86%) as compared to LFD feeding in all three age groups (P<0.0002), with the largest increase again occurring in the young mice (Fig. 1A). The increase in body weight in 6-month old mice on either diet was significantly greater than that in the 12-month old mice, but did not reach statistical significance as compared to the 22-month old mice. All three groups of mice therefore had a marked weight gain that was not significantly affected by aging.
Fig. 1.
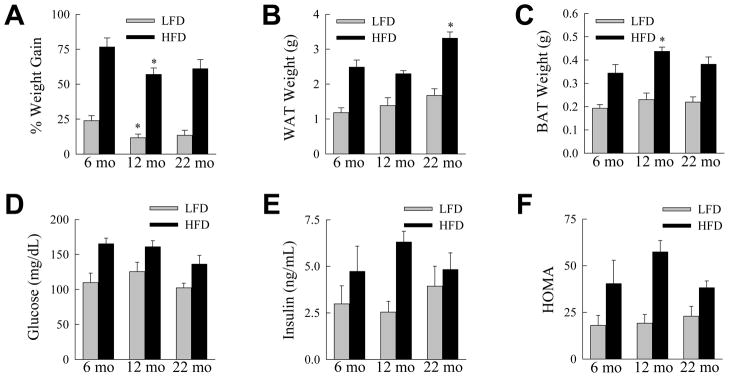
Aging does not affect HFD-induced increases in body and adipose mass and insulin resistance. (A) Percentage weight gain from 16 weeks of LFD or HFD feeding in mice sacrificed at the indicated ages (*P<0.02 as compared to 6-month old mice fed the same diet; n=5–10). (B) WAT weights from the same animals (*P<0.01 as compared to HFD-fed, 6-month old mice; n=5–10). (C) BAT weights (*P<0.03 as compared to HFD-fed, 6-month old mice; n=5–10). (D) Serum glucose levels (n=5–10). (E) Serum insulin levels (n=5). (F) HOMA values (n=5).
The effect of aging on white adipose tissue (WAT) mass was examined as WAT-produced factors contribute to NAFLD development.24 Epididymal WAT mass in LFD-fed mice increased with age but the differences did not reach statistical significance (Fig. 1B). Significant increases in WAT mass did occur with HFD feeding in all three age groups (P<0.001), and the increase was significantly greater in the 22-month old animals (Fig. 1B). Highly metabolic brown adipose tissue (BAT), which may be protective against obesity and its complications,25 was unaffected by aging in LFD-fed animals (Fig. 1C). BAT mass increased significantly in all age groups with HFD feeding (P<0.005), with the increase in 12-month mice being significantly greater (Fig. 1C).
Hyperglycemia and hyperinsulinemia occurred with HFD feeding in all three age groups. Serum glucoses were significantly increased (P<0.03) in HFD-fed as compared to LFD-fed mice at all ages (Fig. 1D). Although serum insulins were significantly elevated in each HFD-fed age group, the difference from the LFD-fed mice was only significant at 12 months (P<0.004; Fig. 1E). Relative insulin sensitivity was determined by HOMA. HOMA values were increased in all HFD-fed groups of mice although none reached statistical significance over LFD-fed mice (Fig. 1F). Thus, the degree of metabolic abnormalities such as obesity and insulin insensitivity resulting from HFD feeding were relatively uniform among young, middle-aged and old mice.
Steatohepatitis but not Simple Steatosis Is Increased with Aging
The effects of aging were examined on the development of NAFLD, the hepatic manifestation of the metabolic syndrome. Liver weight increased significantly (P<0.02) in all age groups with HFD feeding, and the increase was significantly greater in 12- and 22-month old mice (Fig. 2A). However, the increases in liver weight with a HFD paralleled those in whole body mass and the liver to body weight ratios were not significantly changed with diet or aging (Fig. 2B). Hepatic TG content increased significantly (P<0.02), but to an equivalent extent in all three age groups with HFD feeding (Fig. 2C). This biochemical finding was confirmed by blinded grading of the histological degree of steatosis which similarly demonstrated significantly increased steatosis with HFD feeding for all ages (P<0.01), but no significant difference among the three age groups (Fig. 2D). Thus, levels of hepatic steatosis induced by 16 weeks of HFD were equivalent irrespective of age, consistent with the metabolic findings of equal body weight gain and levels of insulin insensitivity in these animals.
Fig. 2.

The development of HFD-induced steatosis is not affected by aging. (A) Liver weights in LFD- and HFD-fed mice sacrificed at the indicated ages (*P<0.05 as compared to HFD-fed, 6-month old mice; n=5–10). (B) Liver weight to body weight ratios in the same mice (n=5–10). (C) Hepatic triglyceride content normalized to liver weight (n=5–10). (D) Histological grade of steatosis (n=5–6).
The effect of aging on steatohepatitis was also assessed. Serum ALT levels were significantly elevated by HFD feeding in the 12- and 22-month old mice (P<0.02), but not in the 6-month old group (Fig. 3A). ALT levels in the HFD-fed older mice were also significantly increased as compared to 6-month old HFD-fed animals (Fig. 3A). As an additional measure of hepatic injury the numbers of TUNEL positive cells in the livers were determined. With HFD feeding the numbers of TUNEL-positive cells increased significantly in all three age groups (P<0.001; Fig. 3B). However, the numbers of TUNEL-positive cells in the 12- and 22-month age groups were significantly increased over the level in the 6-month old HFD-fed animals (Fig. 3B and Fig. 3C-3F). Additional evidence of increased apoptosis was provided by the finding of the active, cleaved forms of the effector caspases 3 and 7 in the livers of 12- and 22-month old HFD-fed mice by immunoblotting (Fig. 4). Thus, despite similar metabolic abnormalities and an equivalent degree of steatosis in the three age groups, significant liver injury was restricted to the older mice.
Fig. 3.
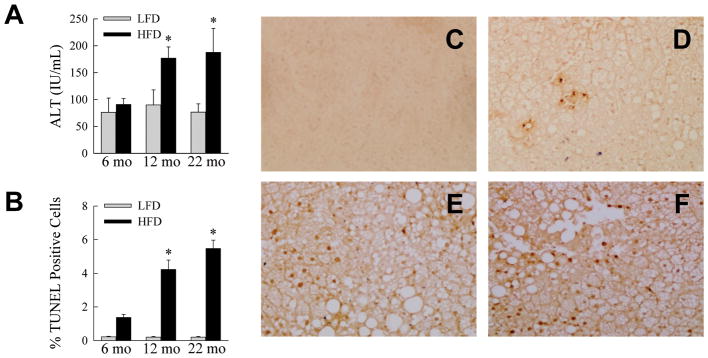
Aging amplifies HFD-induced liver injury. (A) Serum ALTs in LFD- and HFD-fed mice sacrificed at the ages shown (*P<0.03 as compared to HFD-fed, 6-month old mice; n=5–10). (B) Percentage of TUNEL-positive hepatocytes in the livers of LFD- and HFD-fed mice (*P<0.003 as compared to HFD-fed, 6-month old mice; n=4). (C–F) Images of TUNEL-stained cells from a LFD-fed 6-month old mouse (C), a HFD-fed 6-month old mouse (D), a HFD-fed 12-month old mouse (E) and a HFD-fed 22-month old mouse (F).
Fig. 4.
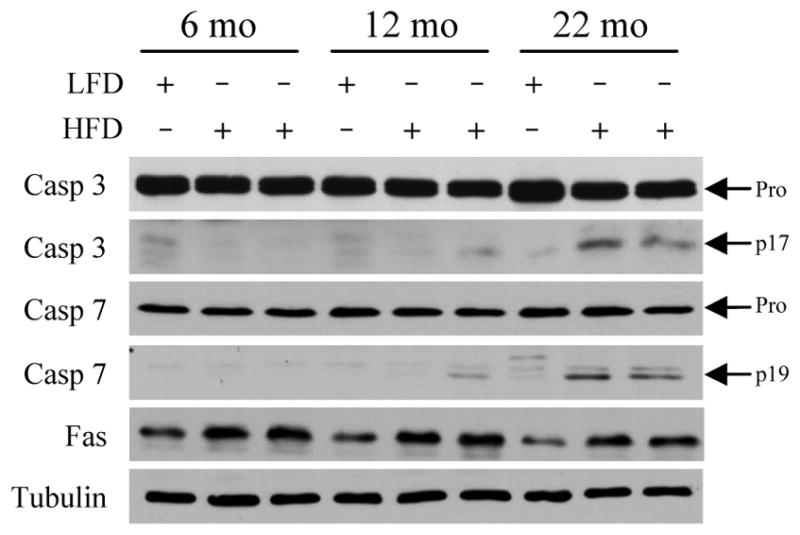
Effector caspases are activated in the livers of aged HFD-fed mice. Immunoblots of total hepatic protein from LFD- and HFD-fed mice for caspase 3 (Casp 3), caspase 7 (Casp 7), Fas and tubulin as a loading control. Arrows indicate the procaspase (Pro) and the cleaved caspase 3 (p17) and 7 (p19) forms.
Aging Promotes a Proinflammatory Immune Response to HFD Feeding
Inflammation in both liver and adipose tissue mediates NAFLD development,26 suggesting that the ability of aging to promote a hyperresponsive inflammatory reaction may underlie the liver injury in older mice. The effects of aging on hepatic inflammation were assessed by relative levels of proinflammatory cytokine gene expression in whole liver. Hepatic levels of TNF mRNA were not significantly increased with HFD feeding as compared to age-matched, LFD-fed mice because of increased TNF levels in older animals on LFD (Fig. 5A). However, TNF levels were significantly increased in the HFD-fed 12- and 22-month old mice as compared to 6-month old HFD-fed mice (Fig. 5A). Levels of MCP-1, a second proinflammatory cytokine implicated in NASH pathogenesis,27 increased with HFD feeding in all ages but were not significantly different from LFD-fed mice or among ages (Fig. 5B).
Fig. 5.
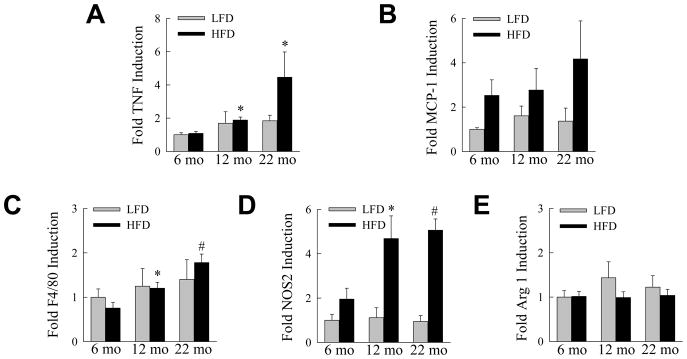
Aging increases hepatic inflammation in response to a HFD. LFD- and HFD-fed mice were sacrificed at the ages shown and mRNA levels in whole liver determined by real time PCR for (A) TNF, (B) MCP-1, (C) F4/80, (D) NOS2 and (E) arginase 1 (*P<0.03, #P<0.002 as compared to HFD-fed, 6-month old mice; n=5–6).
To assess the effect of aging on activation of the hepatic macrophage innate immune response by HFD feeding, levels of the macrophage-specific marker F4/80 were determined in whole liver. Similar to the TNF findings, no significant change occurred between LFD- and HFD-fed mice of the same age, but levels of F4/80 were significantly higher in HFD-fed, older mice as compared to young mice fed the HFD (Fig. 5C). The macrophage response in older mice was also shifted to a more proinflammatory M1 polarized phenotype as levels of the M1 marker NOS228 were significantly increased in the 12- and 22-month old HFD-fed mice (Fig. 5D). In contrast, the M2 marker arginase 128 was unaffected by diet or age (Fig. 5E). Along with the presence of liver injury, aged mice also had an increased M1 proinflammatory macrophage response in the liver.
Adipose tissue was similarly examined for an effect of aging on the immune response to the HFD. TNF levels in epididymal WAT were significantly increased (P<0.05) in all three age groups with HFD feeding, but there was no significant difference among HFD-fed mice of different ages (Fig. 6A). Adipose tissue MCP-1 mRNA levels also increased significantly (P<0.01) but equivalently in all three ages with HFD feeding (Fig. 6B). Consistent with the findings for these two proinflammatory cytokines, F4/80 mRNA levels were significantly and equally increased (P<0.01) in all HFD-fed groups (Fig. 6C). However, significant increases in NOS2 with HFD feeding only occurred in the aged mice (Fig. 6D), whereas significant increases in adipose arginase 1 levels were present in all ages (Fig. 6E). These findings indicate that similar to the liver, WAT in aged mice had a more pronounced M1 macrophage phenotype.
Fig. 6.
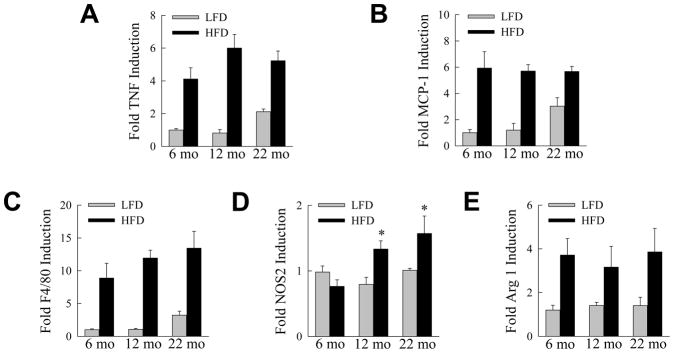
Aging promotes an M1 macrophage phenotype in adipose tissue. mRNA levels in white adipose tissue from LFD- and HFD-fed mice of the indicated ages for (A) TNF, (B) MCP-1, (C) F4/80, (D) NOS2 and (E) arginase 1 (*P<0.01 as compared to HFD-fed, 6-month old mice; n=5–6).
Aged Hepatocytes are Selectively Sensitized to Fas-induced Cell Death
In vivo studies demonstrated that aging sensitized hepatocytes to liver injury in the setting of steatosis in association with an increased inflammatory response. Prominent factors implicated in steatotic liver injury and inflammation include oxidant stress,17 TNF,29 saturated free fatty acids30 and FasL.31,32 To examine whether aging may affect hepatocellular injury from any of these factors, we examined the sensitivity of cultured primary hepatocytes from young (2 month) and aged (18 month) mice to cell death from the oxidant menadione, TNF, the saturated fatty acid palmitate and the Fas agonist antibody Jo2. Primary hepatocytes from young and old mice had equivalent amounts of cell death in response to superoxide-mediated oxidant stress from menadione (Fig. 7A). Hepatocytes from both ages of mice were resistant to cytotoxicity from TNF alone, and equally sensitive to death from actinomycin D and TNF cotreatment (Fig. 7B). Cell death from the toxic saturated free fatty acid palmitate was also equivalent between the two ages (Fig. 7C). In contrast to the findings with these three agents, aged hepatocytes were significantly more sensitive to death from the Fas agonist antibody Jo2 (Fig. 7D). To confirm that aged cells were sensitized to Fas-mediated death, the steady-state levels of apoptosis and necrosis after Jo2 treatment were determined by fluorescence microscopy of acridine orange/ethidium bromide costained cells. Death was primarily apoptotic, and aged hepatocytes had significantly increased numbers of apoptotic and necrotic cells in response to Jo2 (Fig. 7E). To determine whether aging may have additionally sensitized the livers to Fas-mediated injury by up regulating the levels of FasL or its receptor, levels of FasL and Fas mRNA in whole liver were examined by real time PCR in HFD-fed mice. No significant change occurred in FasL or Fas mRNA content in liver with aging (Fig. 8A and 8B). Levels of Fas protein did increase in the liver with HFD feeding, as previously reported for mice fed a high carbohydrate diet,31 but to a similar extent among the three ages of mice (Fig. 4). A possible mechanism for the development of hepatitis in older animals is therefore increased sensitivity of aged hepatocytes to Fas-dependent cell death.
Fig. 7.
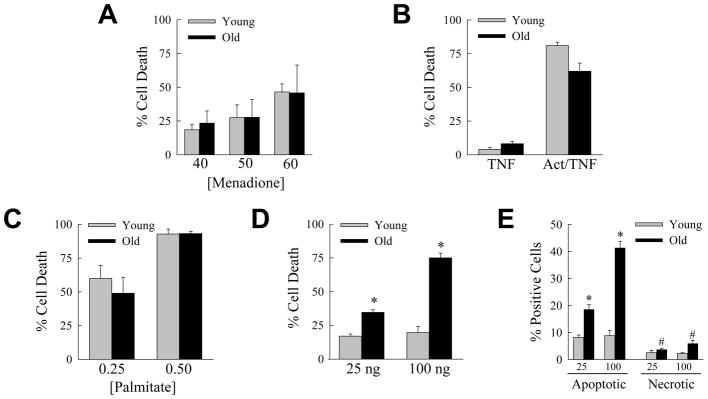
Aged hepatocytes are selectively sensitized to death from the Fas death pathway. (A) Primary hepatocytes were isolated from young (2 month) and old (18 month) mice, cultured and the percentage cell death determined by MTT assay 24 h after treatment with the indicated μM concentrations of menadione (n=7–11). (B) Percentage cell death by MTT assay 24 h after treatment with TNF alone, or cotreatment with actinomycin D and TNF (Act/TNF) (n=4–8). (C) Percentage cell death after 24 h treatment with 0.25 or 0.5 mM palmitate (n=7–8). (D) Percentage cell death by MTT assay 24 h after treatment with the indicated amounts (ng/ml) of Jo2 antibody (*P<0.0001; n=4–8). (E) Percentage of acridine orange/ethidium bromide costained cells that were apoptotic or necrotic by immunofluorescence 12 h after treatment with 25 or 100 ng/ml of Jo2 antibody (*P<0.005, #P<0.03 as compared to young mice; n=3).
Fig 8.

Hepatic FasL and Fas mRNA levels are unaffected by aging. (A) Relative FasL mRNA levels in whole liver from LFD- and HFD-fed mice (n=5–6). (B) Fas mRNA levels in the same livers (n=5–6).
Discussion
Prevalence of the metabolic syndrome increases dramatically with aging.3 Studies have also suggested that aging is associated with an increased prevalence of NAFLD, and in particular that the presence of steatohepatitis and mortality are increased in aged individuals with this disease.4–12 This association may result from age-related increases in obesity and diabetes, cumulative effects on the liver of lifestyle factors such as diet, or the physiological changes that are part of aging. The present study examined these possibilities by determining whether aging affected NAFLD development in mice caused by a defined period of HFD feeding. Interestingly, features of the metabolic syndrome such as increased body mass and insulin insensitivity that developed from HFD feeding were equivalent in young, middle-aged and old mice. Thus, the physiological changes of aging failed to accelerate or worsen the development of two critical components of the metabolic syndrome, obesity and insulin resistance. These findings suggest that rather than age-related changes promoting the development of these metabolic abnormalities, it may be the cumulative length of time or frequency of exposure to factors such as a high-fat diet that increase the prevalence of obesity and insulin insensitivity with aging.
Similar to the findings for obesity and insulin insensitivity, the degree of hepatic steatosis induced by the HFD was unaffected by age. This finding is not surprising in light of the similar extent of weight gain and insulin resistance among the ages which are major risk factors for the development of steatosis. These two factors are thought to play a critical role in the excessive hepatic lipid accumulation that characterizes NAFLD by delivering increased amounts of serum free fatty acids to the liver which are then converted into excessive lipid stores. In contrast, significant steatohepatitis only occurred in older mice demonstrating that aging does play a prominent role in liver injury. Significant hepatocyte injury and cell death as determined by increased ALTs and TUNEL positivity only occurred in the 12- and 22-month old mice. Thus, factors unrelated to the mechanisms of excessive lipid accumulation with aging did promote the development of hepatocellular injury in the setting of steatosis.
Accompanying the liver injury in aged mice was an increased inflammatory response to HFD feeding. This finding is surprising in light of the well-established association of a depressed immune response with aging termed immunosenescence.33 The increase in inflammation may be secondary response to the liver injury. However, findings of a heightened immune response in adipose as well as liver tissue suggest a primary increase in inflammation in response to the HFD in the aged mice. Selective effects were noted with an increase in hepatic TNF but not MCP-1 levels with aging, and a generalized increase in both cytokines in the adipose tissue of all HFD-fed mice irrespective of age. A consistent finding in both tissues was an increased macrophage polarization to a proinflammatory M1 phenotype. This skewed polarization may explain in part the increased inflammatory response in aged animals, and may be a mechanism of liver injury as an unstrained M1 phenotype has been shown to mediate tissue injury in other organs.34
The failure of young mice to progress to steatohepatitis within 16 weeks of HFD feeding contrasts with the increasing prevalence of NASH found in the human pediatric population. Although this inconsistency may simply reflect species differences between mouse and man, alternatively the finding supports the concept that additional factors unrelated to the direct effects of a HFD are necessary to trigger NASH in the young. For example, specific genetic factors may play a critical role in the progression to NASH in young individuals. Increasing evidence suggests that prenatal influences such as maternal imprinting from the effects of obesity and maternal diet promote NAFLD development in young rodents.35,36 The mice employed in the present investigations were the offspring of lean parents fed a normal chow diet and therefore lacked prenatal influences that cause the epigenetic effects necessary for NASH development at a young age. In contrast, in adults other factors may become more critical to promote the progression of HFD-induced steatosis to NASH such as the propensity to develop an adipose tissue inflammatory response.
Cell culture studies of aged hepatocytes suggest that increased hepatocellular injury and death from stimulation of the Fas death pathway may be a mechanism of the increased liver injury in aged mice. Aged hepatocytes did not exhibit a generalized susceptibility to injurious stimuli. Rather aged cells were specifically sensitized to Fas death as they had no increase in death from other possible mediators of liver injury in NAFLD including oxidant stress, TNF or the saturated free fatty acid palmitic acid. Hepatic Fas and FasL mRNA and Fas protein levels were not altered by aging, despite the increase in inflammation, indicating that receptor or ligand up regulation did not contribute to the increase in liver injury and inflammation in aged mice. These findings are to our knowledge the first to identify aging as a modulator of cellular sensitivity to Fas killing, and future studies must attempt to determine the mechanism of this aging effect.
The findings in this study demonstrate that the development of simple steatosis is unaffected by aging presumably because of similar metabolic changes in terms of weight gain and insulin sensitivity that ensue from a HFD irrespective of age. However, physiological changes inherent to aging may promote the development of liver injury and inflammation, and therefore the progression to chronic liver disease and mortality. A further understanding of the mechanisms of these effects may provide new insights into the pathogenesis of NAFLD and lead to specific therapies targeted for elderly individuals with this disease.
Supplementary Material
Abbreviations
- ALT
alanine aminotransferase
- BAT
brown adipose tissue
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HFD
high-fat diet
- HOMA
homeostasis model assessment of insulin resistance
- LFD
low-fat diet
- MCP-1
monocyte chemoattractant protein 1
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NOS2
nitric oxide synthase 2
- TG
triglyceride
- TUNEL
terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling
- TNF
tumor necrosis factor-α
- WAT
white adipose tissue
Footnotes
This work was supported in part by grants DK061498 and AG031782 (MJC), DK041296 and DK020541 from the National Institutes of Health and PR2010-0002 (LF) from the Spanish Ministry of Education (Estancias de Movilidad de Profesores e Investigadores Seniores en Centros Extranjeros de Enseñanza Superior e Investigacion).
Contributor Information
Luis Fontana, Email: fontana@ugr.es.
Enpeng Zhao, Email: ezhao@aecom.yu.edu.
Muhammad Amir, Email: dramir9@gmail.com.
Hanqing Dong, Email: hanqing.dong@gmail.com.
Kathryn Tanaka, Email: ktanaka@montefiore.org.
References
- 1.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 2.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 3.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 4.Lee JY, Kim KM, Lee SG, Yu E, Lim YS, Lee HC, et al. Prevalence and risk factors of non-alcoholic fatty liver disease in potential living liver donors in Korea: a review of 589 consecutive liver biopsies in a single center. J Hepatol. 2007;47:239–244. doi: 10.1016/j.jhep.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Amarapurkar D, Kamani P, Patel N, Gupte P, Kumar P, Agal S, et al. Prevalence of non-alcoholic fatty liver disease: population based study. Ann Hepatol. 2007;6:161–163. [PubMed] [Google Scholar]
- 6.Hamaguchi M, Kojima T, Takeda N, Nakagawa T, Taniguchi H, Fujii K, et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med. 2005;143:722–728. doi: 10.7326/0003-4819-143-10-200511150-00009. [DOI] [PubMed] [Google Scholar]
- 7.Chen CH, Huang MH, Yang JC, Nien CK, Yang CC, Yeh YH, et al. Prevalence and etiology of elevated serum alanine aminotransferase level in an adult population in Taiwan. J Gastroenterol Hepatol. 2007;22:1482–1489. doi: 10.1111/j.1440-1746.2006.04615.x. [DOI] [PubMed] [Google Scholar]
- 8.Kichian K, McLean R, Gramlich LM, Bailey RJ, Bain VG. Nonalcoholic fatty liver disease in patients investigated for elevated liver enzymes. Can J Gastroenterol. 2003;17:38–42. doi: 10.1155/2003/268528. [DOI] [PubMed] [Google Scholar]
- 9.Frith J, Day CP, Henderson E, Burt AD, Newton JL. Non-alcoholic fatty liver disease in older people. Gerontology. 2009;55:607–613. doi: 10.1159/000235677. [DOI] [PubMed] [Google Scholar]
- 10.Argo CK, Northup PG, Al-Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol. 2009;51:371–379. doi: 10.1016/j.jhep.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 11.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J Hepatol. 2008;49:608–612. doi: 10.1016/j.jhep.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 14.Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:159–166. doi: 10.1586/egh.11.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czaja MJ. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implications. Am J Physiol Cell Physiol. 2010;298:C973–978. doi: 10.1152/ajpcell.00527.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 17.Chalasani N, Deeg MA, Crabb DW. Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:1497–1502. doi: 10.1111/j.1572-0241.2004.30159.x. [DOI] [PubMed] [Google Scholar]
- 18.D’Ambra R, Surana M, Efrat S, Starr RG, Fleischer N. Regulation of insulin secretion from β-cell lines derived from transgenic mice insulinomas resembles that of normal β-cells. Endocrinology. 1990;126:2815–2822. doi: 10.1210/endo-126-6-2815. [DOI] [PubMed] [Google Scholar]
- 19.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Jones BE, Neufeld DS, Czaja MJ. Glutathione modulates rat and mouse hepatocyte sensitivity to tumor necrosis factor toxicity. Gastroenterology. 1998;115:1229–1237. doi: 10.1016/s0016-5085(98)70095-2. [DOI] [PubMed] [Google Scholar]
- 21.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, et al. NF-κB inactivation converts a hepatocyte cell line TNF-α response from proliferation to apoptosis. Am J Physiol. 1998;275:C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Xu Y, Nguyen Q, Novikoff PM, Czaja MJ. Induction of hepatoma cell apoptosis by c-myc requires zinc and occurs in the absence of DNA fragmentation. Am J Physiol. 1996;270:G60–G70. doi: 10.1152/ajpgi.1996.270.1.G60. [DOI] [PubMed] [Google Scholar]
- 24.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 25.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Maher JJ, Leon P, Ryan JC. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology. 2008;48:670–678. doi: 10.1002/hep.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 30.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol. 2003;39:978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 32.Siebler J, Schuchmann M, Strand S, Lehr HA, Neurath MF, Galle PR. Enhanced sensitivity to CD95-induced apoptosis in ob/ob mice. Dig Dis Sci. 2007;52:2396–2402. doi: 10.1007/s10620-006-9148-7. [DOI] [PubMed] [Google Scholar]
- 33.Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int. 2009;22:1041–1050. doi: 10.1111/j.1432-2277.2009.00927.x. [DOI] [PubMed] [Google Scholar]
- 34.Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985–997. doi: 10.1172/JCI44490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. doi: 10.1002/hep.23205. [DOI] [PubMed] [Google Scholar]
- 36.Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS One. 2011;6:e21662. doi: 10.1371/journal.pone.0021662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.