

Abstract
Type 1 interferon (IFN) continues to be the foundation for the current standard of care combination therapy for chronic hepatitis C virus (HCV) infection, yet the component interferon‐stimulated genes (ISGs) that mediate the antiviral actions of IFN are not fully defined. Interferon‐induced transmembrane protein 1 (IFITM1) is an ISG product that suppresses early stage infection by a number of viruses through an unknown mechanism of action. Moreover, the actions of IFITM1 on HCV infection are not fully elucidated. Here we identify IFITM1 as a hepatocyte tight junction protein and a potent anti‐HCV effector molecule. IFITM1 expression is induced early during IFN treatment of hepatocytes and accumulates at hepatic tight junctions in HCV‐infected human patient liver during IFN therapy. Additionally, we found that IFITM1 interacts with HCV coreceptors, including CD81 and occludin, to disrupt the process of viral entry. Thus, IFITM1 is an anti‐HCV ISG whose actions impart control of HCV infection through interruption of viral coreceptor function. Conclusion: This study defines IFITM1 as an ISG effector with action against HCV entry. Design of therapy regimens to enhance IFITM1 expression should improve the virologic response among HCV patients undergoing treatment with type I IFN. (Hepatology 2013)
Abbreviations.
HCV, hepatitis C virus; HIV, human immunodeficiency virus; IFITM1, interferon‐induced transmembrane protein 1; IFN, interferon; ISG, interferon stimulated gene; MOI, multiplicity of infection; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; shRNA, short hairpin RNA.
Hepatitis C virus (HCV) is a major public health problem, with over 170 million people worldwide chronically infected.1 HCV therapy has traditionally consisted of interferon (IFN) α and ribavirin. The newly approved protease inhibitors have improved sustained virologic response rates but still require IFN in combination for optimal results and reduced development of viral resistance.2 IFN treatment induces the expression of hundreds of interferon‐stimulated genes (ISGs). ISG products impart immunomodulatory, metabolic, apoptotic, and antiviral actions locally in the infected cell and systematically to bystander cells and immune effector cells, thus mediating antiviral states to suppress infection.3 While the antiviral actions of IFN against HCV are likely mediated by a combination of ISGs acting on multiple viral and host targets, only a few ISGs have so far been defined to have anti‐HCV properties.4, 5, 6, 7, 8, 9, 10 Defining key ISG effectors and their mechanisms of action against HCV will allow for treatment modification to enhance the antiviral activity of IFN‐based therapy toward improved sustained virologic response while also generating potential targets for novel antiviral therapy applications.
IFN triggers ISG expression by binding the interferon α/β receptor and stimulating the Jak/STAT signaling cascade leading to formation of the ISGF3 transcription factor complex and downstream induction of ISGs.11 Different IFN subtypes bind the receptor with varying affinities, leading to different ISG induction profiles.12 We previously showed that the synthetic consensus IFN, a consensus of multiple IFN‐α subtypes, has higher potency against HCV infection compared with naturally occurring IFN‐α subtypes.13 High‐throughput genomics analyses of hepatocytes treated with consensus IFN revealed that this increased potency is associated with the induction of a specific bioset of 86 ISGs, including interferon‐induced transmembrane protein 1 (IFITM1); thus linking IFITM1 to enhancement of IFN antiviral actions.13
IFITM1 and the related IFITM2 and IFITM3 proteins have recently been found to inhibit a number of viruses, including West Nile virus, influenza A, and human immunodeficiency virus (HIV).14,15 A recent study showed an inhibition of HCV replication but not entry by IFITM1, although the mechanism was unclear.16 IFITM1 is induced by type I and type II IFNs17 and has been suggested to mediate the antiproliferative effects of type II IFN.18 IFITM1 has been best characterized in the context of B cells, in which it serves to reduce the threshold of B cell receptor engagement needed for cell stimulation in conjunction with interactions involving CD81 and the B cell receptor.19 Because CD81 is an essential coreceptor for HCV,20 these observations suggest that IFITM1 may impose restriction of HCV infection at the regulation of CD81 function.
HCV enters the host cell through a coordinated pathway of sequential coreceptor interactions that has not been entirely elucidated. HCV is thought to bind heparin sulfate glycoaminoglycans and the LDL receptor at the sinusoidal surface of the hepatocyte as the virus passes through the bloodstream before interacting with the CD81 and SB‐RI co‐receptors, which are engaged by the viral E1/E2 glycoproteins.20 Although claudin‐1 is considered a tight junction molecule, its interactions with CD81 have been shown to occur at the sinusoidal membrane.21 The hepatocyte tight junction is inaccessible to HCV directly in polarized cells,22 and the mechanism by which HCV accesses coreceptors within the tight junction complex remains controversial. The complex orchestration of HCV coreceptors during HCV binding and entry suggests that dysregulation of the coreceptor interaction process offers a prime therapeutic strategy to suppress HCV infection.
Here, we identify IFITM1 as a component of the hepatocyte tight junction and show that IFN‐induced expression of IFITM1 alters the steady state interactions of the HCV coreceptors CD81 and occludin to inhibit HCV entry. Our results identify IFITM1 as an innate immune effector gene that imparts antiviral actions of IFN against HCV through the novel action of disrupting viral coreceptor interactions.
Patients and Methods
Cells and Virus.
Huh7, Huh7‐HCV K2040 subgenomic Con1 (genotype 1b) replicon cells, and PH5CH8 immortalized hepatocyte cells were cultured using standard techniques.23 A Huh7 clone stably expressing FLAG‐IFITM1 and another expressing empty pRev vector were selected and maintained by selection in hygromycin (100 μg/mL) and G418 (100 μg/mL). Huh7 cells stably transfected with IFITM1‐specific short hairpin RNA (shRNA) (TRCN0000057502; Sigma, St. Louis, MO) or nontargeting vector shRNA (SHC002; Sigma) were maintained in complete Dulbecco's modified Eagle's medium containing 2 μg/mL puromycin. An HCV JFH‐1 genotype 2a infectious clone was produced from synthetic complementary DNA constructed from the published sequence of original JFH‐1 clone.24 This virus, termed HCV‐SJ (synthetic JFH1) was then produced directly from RNA, transfected into Huh7.5 cells, and passaged for culture adaptation. The culture‐adapted HCV‐SJ will be described in a subsequent report. For infection studies, HCV‐SJ was grown in Huh7.5 cells, concentrated 100 times using Centricon 100,000 MW cut‐off filters (Millipore, Billerica, MA) and titered via focus‐forming assay on Huh7.5 cells.
Antibodies.
The following antibodies were used: mouse anti‐IFITM1 (Proteintech, Chicago, IL); rabbit anti‐IFITM1 (FL‐125; Santa Cruz Biotechnology, Santa Cruz, CA); FLAG M2 antibody (Sigma); rabbit and mouse anti–ZO‐1, rabbit anti–claudin‐1, and rabbit and mouse anti‐occludin (Invitrogen, Grand Island, NY); E‐cadherin (HECD‐1; Abcam, Cambridge, MA); anti‐CD81 (mouse JS81, BD Pharminogen, San Diego, CA; rabbit H‐121 and mouse 5A6, Santa Cruz Biotechnology); and mouse anti‐HCV NS5a 9E10 (a gift from C. Rice).
Statistical Analysis.
P values were calculated using an unpaired Student t test.
Results
IFITM1 Expression Correlates With Enhanced Anti‐HCV Response.
IFITM1 was identified as a potential anti‐HCV effector molecule through a functional genomics screen of ISGs that direct the enhanced antiviral actions of consensus interferon compared with the naturally occurring IFN‐α 2a and 2b subtypes.13 We validated the enhanced induction of IFITM1 by consensus IFN compared with IFN‐α2a via immunoblot analysis (Fig. 1A). We also assessed IFITM1 expression in vivo in liver tissue from HCV‐infected patients. After 24 hours of IFN‐α therapy, high levels of IFITM1 induction in liver tissue from chronically infected patients correlated strongly with favorable therapeutic outcome of viral load suppression (Fig. 1B). The highest induction of IFITM1 was detected in patients with rapid virologic response to combination pegylated IFN and ribavirin therapy (rapid virologic response: undetectable serum HCV RNA by week 4 of therapy), followed by those with early virologic responses (complete early virologic response: undetectable serum HCV by week 12 of therapy; partial early virologic response: greater than 2 log10 IU/mL reduction in but not complete loss of HCV RNA by week 12 of therapy). Nonresponders were patients in which HCV serum RNA dropped by less than 2 log10 IU/mL by week 12 and was still detectable by week 24 of therapy. IFITM2 and IFITM3 are family members of IFITM1 that are highly related to each other but divergent at the N and C termini from IFITM1.25 IFITM2 and IFITM3 were also induced in rapid virologic response and early virologic response patients but not nonresponders, although we focused this study on the effects of IFITM1. Thus, IFITM1 expression correlates with reduced HCV infection and favorable therapeutic outcome following IFN treatment.
Figure 1.

IFITM1 expression associates with effective anti‐HCV responses. (A) Huh7 cells were treated with CIFN or IFN‐α2a for 0, 8, 16, or 24 hours. IFITM1 protein expression was detected via immunoblot assay. (B) RNA was harvested from HCV‐infected patients 24 hours after IFN‐α therapy. Patients were categorized as rapid virologic responders (RVR; undetectable serum HCV RNA by week 4 of IFN therapy), complete or partial early virologic responders (cEVR and pEVR, respectively; undetectable serum HCV RNA or > 2 log10 IU/mL reduction by week 12 of IFN therapy, respectively) or nonresponders (NR; < 2 log10 IU/mL reduction by week 12 of IFN therapy and serum HCV RNA still detectable by week 24) based on level of HCV control by IFN therapy. Quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) was used to compare IFITM1 levels before and after therapy among these groups.
IFITM1 Mediates Antiviral Actions of IFN Against HCV.
To directly assess the effects of endogenous IFITM1 on HCV replication in Huh7 cells, we knocked down IFITM1 expression by stably expressing an shRNA that targets IFITM1. In the absence of IFN treatment, basal expression of IFITM1 protein was reduced by 99% in the knockdown cells compared with Huh7 control cells stably expressing a nontargeting shRNA construct, and IFITM1 protein levels remained more than 90% reduced even after 24 hours of IFN treatment (Fig. 2A). In the absence of IFN, the knockdown of IFITM1 expression had no effect on HCV infection (Fig. 2B). However, following IFN‐β treatment, HCV RNA levels were significantly increased in IFITM1 knockdown cells compared with control cells (P = 0.0015). Any detectable effect of knocking down a single ISG is notable, as several hundred additional ISGs continue to be expressed following IFN treatment, many exerting independent antiviral functions. These results demonstrate that IFITM1 has antiviral activity and contributes to the global antiviral IFN response during HCV infection.
Figure 2.
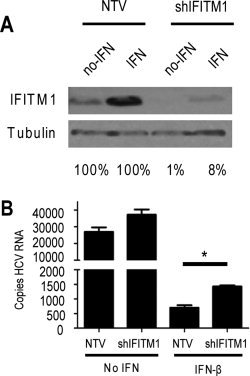
Endogenous IFITM1 contributes to anti‐HCV effects of IFN. (A) Huh7 cells were stably selected for expression of an IFITM1‐specific or nontargeting control shRNA construct. Extracts of cells treated with or without 50 U/mL IFN‐β were probed for IFITM1 expression. Percentages below blots indicate protein levels of IFITM1 relative to nontargeting controls (B) IFITM1 shRNA and nontargeting control Huh7 cells were mock‐treated or treated with 50 U/mL IFN‐β for 16 hours before or 2 hours after HCV infection. Cells were infected with HCV (multiplicity of infection [MOI] = 1), and total RNA harvested at 24 hours postinfection. HCV RNA copy number was quantitated via qRT‐PCR. *P < 0.05.
Mechanism of IFITM1 Anti‐HCV Activity.
To further assess the anti‐HCV effects of IFITM1, we tested the effect of overexpressed IFITM1 on HCV infection. Confocal microscopy analysis of cultures transiently expressing N‐terminally FLAG‐tagged IFITM1 and subsequently infected with culture‐adapted HCV (genotype 2A) revealed a “virus exclusion” phenotype, wherein HCV (as marked by viral protein presence) is effectively excluded from cells expressing IFITM1 but present in neighboring cells lacking IFITM1 expression (Fig. 3A, top panel). In contrast, IFITM1 expression in transfected Huh7‐K2040 cells stably harboring an HCV subgenomic replicon did not exclude viral protein expression (Fig. 3A, lower panel). Since IFITM1 blocks de novo HCV infection but has no detectable effect on the HCV replicon, our data indicate that IFITM1 primarily impacts the early stages of HCV infection. For more detailed analyses, we produced clonal Huh7 cells stably expressing FLAG‐tagged IFITM1 (Huh7‐IFITM1 cells). IFITM1 expression in these cells is four‐fold higher than in Huh7 cells treated with 100 IU/mL IFN‐β for 24 hours (Supporting Fig. 1A). Huh7‐IFITM1 cells were subsequently infected with HCV and assessed for viral RNA accumulation. Constitutive IFITM1 expression in Huh7 cells associated with significantly reduced levels of HCV RNA in infected cultures compared with Huh7 cells stably expressing FLAG vector alone (control) (Fig. 3B). This effect was also seen at the levels of viral protein accumulation (Fig. 3C) and de novo infectious virus production (Fig. 3D). The effect of IFITM1 was not due to defects in a single clone of IFITM1‐expresing Huh7 cells, as a bulk population of Huh7‐IFITM1 also demonstrates reduced HCV protein accumulation (Supporting Fig. 1B). We next tested HCV focus formation on IFITM1‐expressing cultures using confocal microscopy. As expected, IFITM1 expression limited the total number of HCV‐infected foci (Supporting Fig. 1C). Interestingly, for the foci that did form on Huh7‐IFITM1 cells, fewer cells were infected per focus compared with those in vector‐control samples (Figs. 3E, F). This result indicates that HCV is impaired in its ability to spread to neighboring cells in the presence of IFITM1. Together, these data confirm that IFITM1 is a potent IFN‐induced inhibitor of HCV infection.
Figure 3.

Overexpressed IFITM1 inhibits HCV infection. (A) Huh7 cells or Huh7 cells stably harboring the K2040 HCV subgenomic replicon were transiently transfected with FLAG‐tagged IFITM1 followed by HCV infection (MOI = 1). Viral exclusion was assessed by costaining with HCV patient serum (green) and anti‐FLAG (red) followed by confocal microscopy. Nuclei were visualized via DRAQ5 staining. Images were captured at 60× magnification. (B, C) Huh7 cells stably expressing FLAG‐tagged IFITM1 (Huh7‐IFITM1) or vector control (Huh7‐vector) were infected with HCV (MOI = 0.1) and tested for HCV copy number (via qRT‐PCR) (B) and HCV protein levels (determined via immunoblot assay with anti‐HCV NS5a and anti‐HCV patient serum) (C). (D) Huh7‐IFITM1 and Huh7‐vector were infected with HCV (MOI = 1), and supernatants were collected at the indicated times. Viral titers were assessed via focus‐forming assay on Huh7 cells. (E, F) Huh7‐IFITM1 and Huh7‐vector were infected with HCV (MOI = 0.01). Infected cells were assessed by costaining with HCV patient serum (green) and anti‐FLAG (red) followed by confocal microscopy. (E) Representative focus from each cell type. Images were captured at 60× magnification. (F) Number of HCV‐infected cells per focus. Each point represents one focus, and horizontal lines indicate group means. *P < 0.05.
A recent report identified IFITM1 as a restriction factor of HCV replication but not viral entry.16 Although our data do not formally exclude an effect of IFITM1 on HCV replication, they indicate that the primary effect is upstream at the level of binding and/or entry due to the lack of inhibition of HCV replicon. The known interaction of IFITM1 with the HCV coreceptor CD81 suggests that IFITM1 could alter the function of one or more HCV receptors and inhibit the entry process. We determined that pseudoparticles enveloped with HCV glycoproteins were significantly reduced in their ability to enter Huh7‐IFITM1 compared with Huh7‐vector cells (Fig. 4A). In contrast, VSV and MLV pseudoparticles were not significantly reduced in the presence of IFITM1 (Fig. 4A). While this result indicates that IFITM1 reduces HCV entry, the effect is not as strong as expected by the earlier reduction of HCV infection itself. To more carefully examine HCV binding and entry, we tested each stage with actual infectious virus. To determine whether IFITM1 inhibited HCV binding to or entry into host cells, Huh7 cells were incubated with HCV virions at 4°C and thereafter unbound virus was washed away. HCV RNA collected at this point represents HCV bound to the cell surface.26 After virus binding, cells were shifted to 37°C to allow HCV entry to proceed. After 4 hours the HCV virions that remained on the cell surface were stripped off by proteinase K treatment of cells, and therefore the HCV RNA collected at this point represents only cell‐internalized virus. As a negative control of virus binding and entry, we included a parallel assessment of cells treated with neutralizing anti‐CD81 antibody.27 Expression of IFITM1 did not significantly reduce HCV binding, which remained stable and above the levels of virus binding after anti‐CD81 treatment. Thus, IFITM1 does not function to block CD81‐dependent binding of HCV to target cells (Fig. 4B). Importantly, we found that HCV entry was blocked by ectopic IFITM1 expression to a level similar to the blockade imposed by treatment with anti‐CD81 antibody (Fig. 4B). These data indicate that, while HCV can bind to hepatocytes in the presence of IFITM1, the virus is unable to enter cells and complete the infection process. The reduction of HCV entry to levels corresponding to an anti‐CD81 control suggests that CD81‐dependent HCV entry is regulated by IFITM1.
Figure 4.

IFITM1 inhibits HCV entry into hepatocytes. (A) Huh7‐IFITM1 and Huh7‐vector were infected with pseudoparticles containing envelopes of the indicated viruses. Viral entry was determined by luciferase activity in relative light units. (B, C) Huh7‐IFITM1 or Huh7‐vector, each with and without neutralizing anti‐CD81, were bound with HCV (MOI = 1) at 4°C for 1 hour. Cells were either harvested for RNA to assess binding (B) or shifted to 37°C for 5 hours, followed by proteinase K cleavage of external virus and subsequent RNA collection to assess entry (C). HCV copy number was determined via qRT‐PCR. The dotted line on the HCV entry panel refers to the limit of detection for this HCV qRT‐PCR assay. *P < 0.05.
IFITM1 Associations with HCV Coreceptors.
IFITM1 is known to physically interact with CD81, an essential coreceptor of HCV.20,28 We confirmed this interaction in Huh7 cells by immunoprecipitation/immunoblot assay, as well as an association of IFITM1 with occludin, another required HCV coreceptor (Fig. 5A). These immunoprecipitations were performed using equivalent amounts of protein lysates in CHAPS buffer, which has been demonstrated to preserve CD81 interactions with associated proteins.28 We have experimentally determined that these conditions lead to consistent and specific immunoprecipitation reactions. Because occludin is a component of the tight junction in polarized cells (unlike Huh7 cells which are unable to polarize under cell culture conditions29), we were interested in the potential association of IFITM1 with other molecules involved in tight junctions. We found that IFITM1 associates with the tight junction marker ZO‐1, but not the adherens junction marker E‐cadherin30,31 (Fig. 5A). Although these data do not formally define an interaction of IFITM1 with these complexes due to the lack of Huh7 polarization, they demonstrate the potential for such associations in liver cells.
Figure 5.

IFITM1 alters HCV coreceptor associations. (A) Total cell lysates from Huh7‐IFITM1 or Huh7‐vector were immunoprecipitated as indicated and probed via anti‐FLAG immunoblotting to assess IFITM1 pull‐down. (B) Total cell lysates from Huh7‐IFITM1 and control cells, mock‐ and IFN‐β–treated Huh7 cells, and mock‐ and IFN‐β–treated Huh7 cells harboring nontargeting vector (NTV) or IFITM1‐specific shRNA (shIFITM1) were immunoprecipitated with anti‐CD81 and probed by immunoblot assay assessment of occludin and ZO‐1.
The association of IFITM1 with HCV coreceptors suggests that IFITM1 could inhibit HCV infection by disrupting the coordinated sequence of coreceptor associations required for viral entry. This process could include an IFITM1‐mediated disruption of the essential CD81 association with other HCV coreceptors. To determine how IFITM1 impacts one such CD81 interaction, we conducted anti‐CD81 immunoprecipitation/immunoblot analyses in the presence or absence of ectopic IFITM1. Surprisingly, expression of IFITM1 led to a redistribution of CD81 into occludin‐containing complexes (Fig. 5B). Moreover, we found that IFITM1 promoted the association of CD81 with ZO‐1. This result indicates that IFITM1 enhances the association of CD81 with a complex typically associated with tight junctions in polarized cells. We also discovered that IFN treatment of cultured cells resulted in an increased association of CD81 with occludin (Fig. 5B). The enhancement of CD81/occludin binding was dependent on IFITM1 expression, since both basal and IFN‐induced CD81‐occludin associations were reduced in IFITM1‐shRNA knockdown cells (Fig. 5B). Importantly, the enhanced association of CD81 with this complex was independent of HCV infection, as IFITM1 expression induced exogenously or by IFN treatment produced this phenotype in the absence of infection. Thus, IFITM1 expression stimulates CD81 redistribution and a stable association between CD81 and occludin during the IFN response. Alteration of CD81 associations in noninfected cells suggests that IFITM1 might disrupt the process of HCV infection through altered coreceptor function.
IFITM1 Is a Tight Junction Molecule in Hepatocytes.
The apparent association of IFITM1 with occludin and ZO‐1 (Fig. 5A) suggests that IFITM1 might localize to tight junctions in polarized hepatocytes as found in liver tissue. Because we cannot make conclusions about tight junction molecule localizations in Huh7 cells due to their inability to polarize, we assessed IFITM1 distribution in liver tissue biopsies from HCV‐infected patients after 24 hours of IFN therapy (Fig. 6A). We found that IFITM1 was distributed in liver tissue in a pattern consistent with hepatic tight junction localization and fully overlapped with the tight junction marker ZO‐1. While this result demonstrated the tight‐junction localization of IFITM1 in liver tissue, the possibility remained that IFITM1 may move to tight junctions during the course of HCV infection through an interaction with CD81.
Figure 6.

IFITM1 localizes to hepatic tight junctions. (A) Liver tissue from chronically infected HCV patients treated with IFN‐α for 24 hours was costained for IFITM1 (green) and ZO‐1 (red) and visualized using confocal microscopy. (B) Liver tissues from SCID‐uPA chimeric mice treated with IFN‐α for 7 days were costained for IFITM1 in green and tight junctions in red. Samples were visualized using confocal microscopy. (C) Three‐dimensional reconstructions of confocal analysis in panel B. All images were captured at 60× magnification.
We next examined the localization of IFITM1 following IFN treatment of non‐infected liver, using the SCID/Alb‐uPA chimeric mouse. These mice harbor humanized livers and effectively support HCV infection and the innate immune response against HCV.32,33 Noninfected mice were treated with IFN‐α for 7 days, at which time livers were harvested and processed for RNA and protein assessment, as well as analysis of liver sections. We found that IFITM1 expression was induced by IFN in chimeric mouse liver tissue (Supporting Fig. 2). We also conducted confocal microscopy of chimeric liver sections costained for IFITM1 and ZO‐1 or occludin (Fig. 6B). Similar to IFN treatment of patient liver, IFITM1 colocalized with tight junctions in IFN‐treated chimeric mouse liver, demonstrating a tight junction pattern of localization. Colocalization was confirmed by three‐dimensional reconstruction of confocal images (Fig. 6C), demonstrating full overlap of IFITM1 with ZO‐1 and occludin. Thus, IFITM1 localizes to hepatic tight junctions following IFN treatment of uninfected chimeric mice or HCV‐infected patients.
Discussion
The current standard of care for chronic hepatitis C virus infection relies on the antiviral action of type I interferon. IFN functions through the induction of several hundred ISGs; however, the set of antiviral effector ISGs required for IFN‐mediated control of HCV has not yet been elucidated. The failure of the current IFN‐based therapy to achieve sustained viral inhibition in a significant portion of patients treated, coupled with the inability of many patients to tolerate the widespread side effects of IFN therapy, drives the need for more effective and targeted treatments. An attractive strategy to improve the IFN component of current combination therapies includes refining IFN therapy for enhancement of specific antiviral ISG actions and elucidating mechanisms of ISG effector function to identify novel therapeutic targets.
Here, we define IFITM1 as an anti‐HCV effector ISG that inhibits entry of HCV into hepatocytes. We found that IFITM1 localizes to hepatic tight junctions. IFITM1 mediates CD81 relocalization into the tight junction complex to impart an enhanced association with occludin and other tight junction factors. We propose that the anti‐HCV actions of IFITM1 are attributed to its ability to disrupt the coordination of coreceptor interactions. These actions impart control of HCV by disrupting the assembly of HCV coreceptor complexes to suppress the HCV entry process. We conclude that IFITM1 is an important anti‐HCV effector ISG whose actions limit HCV infection through disruption of the viral entry process.
The IFITM family has been shown to impose antiviral actions against a number of viruses, including influenza A virus, SARS coronavirus, West Nile virus and HIV.14,15,34 While these studies also showed that each of these viruses are restricted by IFITM1 at early stages of infection, the viruses studied do not share major viral entry receptors with HCV, nor are they known to involve tight junctions in their entry processes. This raises the question as to how IFITM1 exerts its antiviral function against different viruses. Notably, tetraspanins including CD81 have been shown to regulate HIV‐induced cell fusion, and CD81 was recently identified to be necessary for optimal entry by influenza A virus.35, 36, 37 These reports thus identify a common thread of at least three viruses known to be inhibited by IFITM1 through a class of molecules known to include IFITM1‐interacting partners.28 Tetraspanins have been demonstrated to regulate fusion of a number of additional viruses,38 so it is likely that other IFITM1‐regulated viruses use tetraspanins to regulate viral entry. Our finding that IFITM1 inhibits HCV entry is in contrast to a recent publication that showed that IFITM1 was unable to inhibit cellular entry by HCV pseudoparticles, yet it reduced viral replication.16 This discrepancy is likely due to lower sensitivity of the HCV pseudoparticle system, as demonstrated in Fig. 4. We were also unable to recapitulate the authors′ reported effect of IFITM1 on HCV replication. This can in part be explained by a recent siRNA screen in which IFITM1 siRNA enhances HCV replication in the context of IFN treatment, but IFITM1 overexpression alone had no significant effect.10 We propose that IFITM1 might in part affect HCV replication, but that the primary effect occurs at the level of viral entry, as in the case of other viruses studied. Another interesting component of antiviral activity of IFITM1 would be that against other viruses known to use the tight junction during their entry processes, such as coxsackie virus.39 Our data thus far are not sufficient to make informed hypotheses about these viruses. One could propose that IFITM1 alters complexes involved in tight junctions and may affect these other viruses, or that there may be no effect on other viruses if they do not require affected coreceptors such as CD81 or occludin.
The commonality of CD81 in the entry process of several IFITM1‐regulated viruses provides a hypothesis for the function of IFITM1. Data presented in this study demonstrate that IFITM1 functions at the stage of viral entry, downstream of virus binding to the cell. This has also been demonstrated for HIV, where the virus was able to bind host cells and enter endosomal compartments, but was not released into the cytoplasm in the presence of IFITM1.15 Because CD81 is known to regulate host and viral membrane fusions,38 its complex redistribution in the presence of IFITM1 may alter the CD81‐mediated regulation of viral fusion and subsequent entry into host cells. The primary role of tetraspanins such as CD81 appears to be the organization of membrane complex formation,38 an essential process in viral entry into host cells and a likely target of IFITM1‐mediated viral disruption. The tight junction localization of IFITM1 provides new insights into the cellular functions of this molecule. Strategies that effectively induce IFITM1 expression and function could serve to improve existing anti‐HCV therapies to increase sustained virologic response rates.
Supporting information
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information
Acknowledgements
We thank C. Rice for reagents; M. Brassil, K. Chang and N. Crochet for technical assistance; S. Horner and G. Schnell for discussion and critical reading of the manuscript; and the W. M. Keck Center for Advanced Studies in Neural Signaling for microscopy assistance.
Potential conflict of interest: Dr. Tyrrell owns stock in and holds intellectual property rights with KMT Hepatech.
This work was supported by National Institutes of Health grants AI060389, AI40035, and DA024563 (to M. G.) and DK068598‐01 (to D. T.‐Y. L.) and the Burroughs Wellcome Fund (M. G.).
References
- 1. Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(Suppl. 1): 74‐81. [DOI] [PubMed] [Google Scholar]
- 2. Kieffer TL, Sarrazin C, Miller JS, Welker MW, Forestier N, Reesink HW, et al. Telaprevir and pegylated interferon‐alpha‐2a inhibit wild‐type and resistant genotype 1 hepatitis C virus replication in patients. HEPATOLOGY 2007; 46: 631‐639. [DOI] [PubMed] [Google Scholar]
- 3. Wilkins C, Gale M Jr. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol 2010; 22: 41‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gale M Jr. Effector genes of interferon action against hepatitis C virus. HEPATOLOGY 2003; 37: 975‐978. [DOI] [PubMed] [Google Scholar]
- 5. Wang C, Pflugheber J, Sumpter R Jr, Sodora DL, Hui D, Sen GC, et al. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol 2003; 77: 3898‐3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang D, Guo H, Xu C, Chang J, Gu B, Wang L, et al. Identification of three interferon‐inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol 2008; 82: 1665‐1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Helbig KJ, Eyre NS, Yip E, Narayana S, Li K, Fiches G, et al. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. HEPATOLOGY 2011; 54: 1506‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schoggins JW, Rice CM. Interferon‐stimulated genes and their antiviral effector functions. Curr Opin Virol 2011; 1: 519‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao H, Lin W, Kumthip K, Cheng D, Fusco DN, Hofmann O, et al. A functional genomic screen reveals novel host genes that mediate interferon‐alpha's effects against hepatitis C virus. J Hepatol 2012; 56: 326‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, et al. Identification of type I and type II interferon‐induced effectors controlling hepatitis C virus replication. HEPATOLOGY 2012; doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 11. Joshi S, Kaur S, Kroczynska B, Platanias LC. Mechanisms of mRNA translation of interferon stimulated genes. Cytokine 2010; 52: 123‐127. [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto S, Yano H, Sanou O, Ikegami H, Kurimoto M, Kojiro M. Different antiviral activities of IFN‐alpha subtypes in human liver cell lines: synergism between IFN‐alpha2 and IFN‐alpha8. Hepatol Res 2002; 24: 99. [DOI] [PubMed] [Google Scholar]
- 13. Erickson AK, Gale M Jr. Regulation of interferon production and innate antiviral immunity through translational control of IRF‐7. Cell Res 2008; 18: 433‐435. [DOI] [PubMed] [Google Scholar]
- 14. Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009; 139: 1243‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. The IFITM proteins inhibit HIV‐1 infection. J Virol 2011; 85: 2126‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol 2011; 85: 12881‐12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reid LE, Brasnett AH, Gilbert CS, Porter AC, Gewert DR, Stark GR, et al. A single DNA response element can confer inducibility by both alpha‐ and gamma‐interferons. Proc Natl Acad Sci U S A 1989; 86: 840‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang G, Xu Y, Chen X, Hu G. IFITM1 plays an essential role in the antiproliferative action of interferon‐gamma. Oncogene 2007; 26: 594‐603. [DOI] [PubMed] [Google Scholar]
- 19. Levy S, Todd SC, Maecker HT. CD81 (TAPA‐1): a molecule involved in signal transduction and cell adhesion in the immune system. Annu Rev Immunol 1998; 16: 89‐109. [DOI] [PubMed] [Google Scholar]
- 20. Zeisel MB, Fofana I, Fafi‐Kremer S, Baumert TF. Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies. J Hepatol 2011; 54: 566‐576. [DOI] [PubMed] [Google Scholar]
- 21. Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, et al. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem 2010; 285: 21092‐21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mee CJ, Harris HJ, Farquhar MJ, Wilson G, Reynolds G, Davis C, et al. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J Virol 2009; 83: 6211‐6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG‐I. J Virol 2005; 79: 2689‐2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kato T, Date T, Murayama A, Morikawa K, Akazawa D, Wakita T. Cell culture and infection system for hepatitis C virus. Nat Protoc 2006; 1: 2334‐2339. [DOI] [PubMed] [Google Scholar]
- 25. Martensen PM, Justesen J. Small ISGs coming forward. J Interferon Cytokine Res 2004; 24: 1‐19. [DOI] [PubMed] [Google Scholar]
- 26. Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, et al. CD81 is dispensable for hepatitis C virus cell‐to‐cell transmission in hepatoma cells. J Gen Virol 2009; 90: 48‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cormier EG, Tsamis F, Kajumo F, Durso RJ, Gardner JP, Dragic T. CD81 is an entry coreceptor for hepatitis C virus. Proc Natl Acad Sci U S A 2004; 101: 7270‐7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi S, Doss C, Levy S, Levy R. TAPA‐1, the target of an antiproliferative antibody, is associated on the cell surface with the Leu‐13 antigen. J Immunol 1990; 145: 2207‐2213. [PubMed] [Google Scholar]
- 29. Decaens C, Durand M, Grosse B, Cassio D. Which in vitro models could be best used to study hepatocyte polarity? Biol Cell 2008; 100: 387‐398. [DOI] [PubMed] [Google Scholar]
- 30. Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO‐1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol 1986; 103: 755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin‐based adherens junctions. Annu Rev Cell Dev Biol 1997; 13: 119‐146. [DOI] [PubMed] [Google Scholar]
- 32. Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, et al. Hepatitis C virus replication in mice with chimeric human livers. Nat Med 2001; 7: 927‐933. [DOI] [PubMed] [Google Scholar]
- 33. Walters KA, Joyce MA, Thompson JC, Smith MW, Yeh MM, Proll S, et al. Host‐specific response to HCV infection in the chimeric SCID‐beige/Alb‐uPA mouse model: role of the innate antiviral immune response. PLoS Pathog 2006; 2: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, et al. Distinct patterns of IFITM‐mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 2011; 7: e1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thali M. Tetraspanin functions during HIV‐1 and influenza virus replication. Biochem Soc Trans 2011; 39: 529‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gordon‐Alonso M, Yanez‐Mo M, Barreiro O, Alvarez S, Munoz‐Fernandez MA, Valenzuela‐Fernandez A, et al. Tetraspanins CD9 and CD81 modulate HIV‐1‐induced membrane fusion. J Immunol 2006; 177: 5129‐5137. [DOI] [PubMed] [Google Scholar]
- 37. Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, et al. Human host factors required for influenza virus replication. Nature 2010; 463: 813‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fanaei M, Monk PN, Partridge LJ. The role of tetraspanins in fusion. Biochem Soc Trans 2011; 39: 524‐528. [DOI] [PubMed] [Google Scholar]
- 39. Coyne CB, Bergelson JM. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev 2005; 57: 869‐882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information