

Abstract
The oxygen atom transfer reactivity (OAT) from dioxo-Mo(VI) complexes of hydrotrispyrazolyl borate (hydrotris(3,5-dimethylpyrazolyl)borate, TpMe2; hydrotris(3-isopropylpyrazol-1-yl)borate, TpiPr) to tertiary phosphines (PMe3, PMe2Ph, PEt3, PEt2Ph, PBun3, PMePh2, or PEtPh2) has been investigated. In acetonitrile, these reactions proceed via the formation of a phosphoryl intermediate complex that undergoes a solvolysis reaction. We report the synthesis and characterization of several phosphoryl complexes. The rates of formation of phosphoryl complexes and their solvation were determined by spectrophotometry. The rates of the reactions and the properties of the phosphoryl species were investigated using the Quantitative Analysis of Ligand Effect (QALE) methodology. The results show that, at least in this system, the first step of the reaction is controlled primarily by the steric factor, and in the second step, both electronic and steric factors are important. We also analyzed the effect of ligands on the reaction rate e.g., TpMe2 vs. TpiPr.
Oxygen atom transfer (OAT) reactions are ubiquitous in chemical processes, and involve the breaking of the oxygen-donor bond and the making of the oxygen-acceptor bond. The reactions can be either catalytic or stoichiometric where the source of oxygen can be molecular oxygen, peroxide, or water. In biological systems, when both atoms of molecular oxygen are inserted into the substrates they are called ‘dioxygenase’, and only one oxygen atom is inserted they are called ‘monooxygenase’. Many pterin containing molybdenum enzymes fall under the latter category, as they catalyze the transfer of an oxygen atom to or from the substrate.1, 2 Examples of such enzymes include dimethyl sulfoxide (DMSO) reductase which reduces DMSO to DMS and sulfite oxidase (SO) which oxidizes sulfite to sulfate. In both cases, the molybdenum center shuttles between +6 and +4 oxidation states. In the case of DMSO reductase, the fully oxidized Mo +6 state is coordinated by one terminal oxo ligand that serves as the oxo-donor. In the case of SO, the fully oxidized Mo +6 state is coordinated by two terminal oxo ligands. The physiological reaction of SO i.e., OAT reactivity from a dioxo-Mo center has been investigated to gain insight into details about the reaction pathway. The substrates of choice have been tertiary phosphine (PR3), as its physiochemical properties can be tuned, via substitution at phosphorus. Phosphines are not physiological substrates for any pterin-containing molybdenum enzymes, but they have been used in enzymatic studies such as the catalysis of a water soluble phosphine oxidation by DMSO reductase,3 and hydrophobic PMe3 has been used in a voltammetric investigation of DMSO reductase4.
We have been investigating5–14 the mechanistic details of OAT reactions between tertiary phosphine (R3P) and oxo-Mo(VI) complexes of hydrotrispyrazolyl borate ligands (Tp) as a vehicle to understand the OAT reactivity in enzymatic systems, simultaneously contribute in developing a fundamental understaning in this important chemical reactions.. Specifically, we used dioxo-Mo(VI) complexes of hydrotris(3,5-dimethylpyrazolyl)borate ligand, TpMe2 and hydrotris(3-isopropylpyrazol-1-yl)borate, TpiPr ligands (Scheme 1). It is generally believed that the first step in OAT reactions from dioxo-Mo(VI) centers to tertiary phosphines involves nucleophilic attack of the phosphine P on an empty Mo=O π* orbital, while the other oxo-group acts in a ‘spectator’ capacity.15–17 In this general scheme, we described a reciprocity such that the terminal oxo group also attacks on the phosphine through the P—C σ* orbital.18 We also demonstrated that in some cases, the result is the formation of a phosphine oxide coordinated intermediate, which subsequently, can undergo a solvolysis reaction generating a phosphine oxide and solvent coordinated Mo(IV) center.18 This electronic description implies that the initial reaction is dependent on the nucleophilicity of the reacting phosphine i.e., the higher the nucleophilicity, the faster the rate of reaction. However, the reaction with phosphine is rarely viewed as under complete electronic control. Indeed, we have demonstrated that the reaction rate of the phosphine oxide complex formation varies linearly with the steric properties of the phosphine (ca. the cone angle).5 Such a behavior, however, is not unique to molybdenum complexes.
Scheme 1.
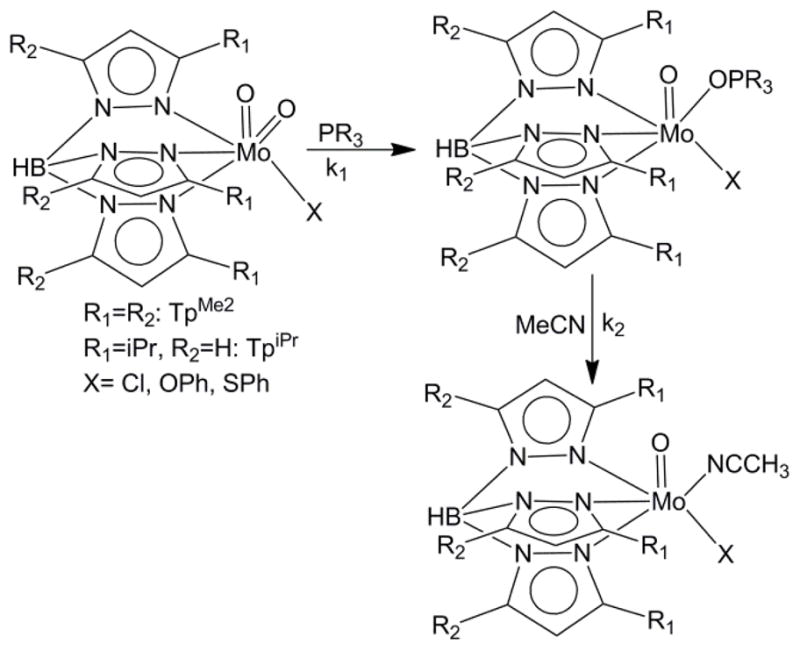
A Two-step model of OAT reaction
In 1966 Basolo and coworkers described a linear free energy relationship between the basicity of a reagent with and the rate of reaction.19 Specifically, the rate of the substitution reaction of CO in CpM(CO)2 varies linearly with the basicity of tertiary phosphines i.e., higher the basicity faster the reaction rate. However, the authors argued that the polarazibility, not the basicity is a better parameter to consider in understanding the reactivity. In another seminal work, Tolman introduced the concept of the ‘phosphine cone angle’ as a measure of the steric interaction of phosphine.20, 21 He also recognized that the CO stretching frequency in Ni(CO)3PR3can be shifted through variable substitution at phosphorous, and proposed a linear relation (eq 1) correlating the property of a complex with electronic and steric effects, where, a
| eq 1 |
is a constant, b and c are weighting factors, θ is the cone angle and χ is Tolman’s electronic parameter as defined by the shift in the νCO (A1) vibration of Ni(CO)3PR3 complexes, referenced to PtBu3 complex.21 Equation 1 provides a means to separate the steric and electronic contributions, which has been subsequently modified, and a general form is shown in eq2.
| eq 2 |
Where, a, b, c, d and e are constants, χd is the σ donor capacity of the reagent – smaller the value, stronger the σ donor capacity (range: −0.9 to 43); θ is the Tolman’s cone angle and θst defines the steric threshold, beyond which the steric effect becomes important; λ is defined as a switching function; Ear is a parameter that accounts for the aryl effects and is dependent on the number of aryl groups attached to the phosphine; πp defines measure of π-acidity. This relation has been used in quantifying contributions to the observed property, and the approach is referred to as ‘Quantitative Analysis of Ligand Effect’ (QALE). The QALE model separates and quantifies different ligand properties such as σ donation ability, π acidity and steric contribution with the understanding that the steric influence may be discontinuous in nature.
The oxygen atom transfer reactivity of dioxo-Mo centers have been investigated for decades.22–59 However, there hasn’t been any in-depth investigation detailing how the stereoelectronic properties of phosphines can impact the reaction. Lorber et al.,60 has reported the second order rate constants for reaction of phosphine with dioxo-Mo(VI) maleonitrile complexes (eq 3) vary linearly with the electronic parameters of the ligand but are almost
| eq 3 |
invariant with the ligand cone angle, which is different than what we observed with the pyrazolyl borate ligand system.5 This prompted us to look into the details of the OAT reactions as a whole in the pyrazolyl borate system. Herein we discuss the QALE analysis of the rate constants of the OAT reactions of TpMoVIO2X (where TpiPrMoVIO2(OPh), 1; TpMe2MoVIO2Cl, 2; TpMe2MoVIO2(OPh), 3; and TpMe2MoVIO2(SPh), 4) with different PR3. This reaction yields the phosphoryl complexes shown in Chart 1.
Chart 1.

In acetonitrile the phosphoryl complexes undergo a solvolysis reaction (Scheme 1) generating TpMoOX(MeCN) (i.e., TpiPrMoO(OPh)(MeCN), 5; TpMe2MoOCl(MeCN), 6; TpMe2MoO(OPh)(MeCN), 7; and TpMe2MoO(SPh)(MeCN), 8. Using the kinetic data we show that both the steric and electronic parameters are important and their relative significance depends on the particular system. For completeness, we also report the syntheses and characterization of several new phosphoryl complexes. In addition, we report their redox potential and 31P chemical shifts and analyses of these data using QALE methodogy.
Experimental
Materials and Methods
All solvents, except the NMR solvents and acetonitrile, were distilled from sodium, deoxygenated using the freeze-pump-thaw technique, and stored inside a glove-box until needed. The acetonitrile used in this study was triple-distilled from KMnO4, CaH2 and then P4O10, and was stored anaerobically. Some of the NMR solvents were dried by flushing them through an activated alumina column inside a glove-box under nitrogen, while others were used as received. In addition, all solutions were prepared under an argon atmosphere using standard Schlenk techniques and gas-tight syringes. The syntheses and characterization of 1,10 2–4,61 1c,10 1f,10 2a–2c,9, 11 4a7, 8, 5,10 6,9 87, 8 have been reported elsewhere.5, 7–11, 14,62 Selected kinetic data (i.e., formation and solvolysis of 1a–1g,5, 18 and 4a7, 8) and methodology for kinetic experiments6, 18 have been reported. NMR spectra of 1–4 were recorded on a Bruker 300 MHz NMR spectrometer either in CD3CN or in C6D6. NMR spectra for the characterization of the intermediates (1a–1g, 2a–2g, 3a–3g, 4a and 4g) were recorded on a Varian 500 MHz spectrometer, with 31P spectra referenced to PPh3 in C6D6 solution. Electronic spectra were recorded on a Cary 14 spectrophotometer with an OLIS 14 (operating system version 2.6.99) connected to a constant-temperature circulator. Electrochemical measurements were carried out in a Bioanalytical Systems (BAS) model CV-50W cyclic voltammeter. Voltammograms were recorded with a standard three-electrode system consisting of a Pt-disk working electrode, a Ag/Ag+ reference electrode, a Pt-wire auxiliary electrode, and tetrabutylammonium perchlorate (TBAP) as the charge carrier.63 All voltammograms were internally referenced with ferrocene, and the potentials are reported with respect to the Fc+/Fc couple without junction correction. In addition, all electrochemical work was performed inside a dry-box, under a nitrogen atmosphere, at room temperature.
TpMe2MoOCl(OPEt2Ph) (2d)
This complex was synthesized following the general procedure described for 2a, 2b and 2c.9, 11 A round bottom flask containing 2 and a stir bar was placed in the dry box, approximately 15–20 mL of benzene was added and the flask was capped and cooled overnight. The flask was returned to the stir plate and an excess (two times) of ice cold PEt2Ph was added and allowed to react for approximately 2 minutes, resulting in a yellowish-green solution. The reaction mixture was filtered, and the unreacted TpMe2MoO2Cl was washed three times (10 mL each) with hexane. The yellowish-green filtrate was immediately evaporated to dryness and washed again three times (10 mL each) with ice cold hexane, resulting in the greenish-yellow TpMe2MoOCl(OPEt2Ph). νMo=O 947 cm−1. νP=O 1090 cm−1. δ1H (ppm) o-HC OPEt2Ph (dd) 7.397 :m-HC OPEt2Ph (dt) 7.005: p-HC OPEt2Ph (dt) 6.916: HC(Pz) (s) 1H each: 5.756, 5.675, 5.503, H3C(Pz) (s) 3H each: 3.022, 2.668, 2.653, 2.247, 2.199, 2.076: H2C OPEt2Ph (m) 2H each 2.740, 2.480, 1.495, 1.159: H3C OPEt2Ph (dt) 0.938, 0.744. δ31P (ppm) 67.403. λmax (dd) 12316 cm−1 (ε, 51 mol−1•cm−1); 22673cm−1 (ε, 112 mol−1•cm−1).
TpMe2MoOCl(OPMePh2) (2f)
This complex was synthesized following the procedure developed for 2d, with the exception that compound 2 was allowed to react less than a minute, and produced a yellow-gold color upon reaction with an excess (two times) of ice cold PMePh2. Compound 2f was isolated as a yellow-gold powder. νMo=O 953 cm−1. νP=O 1090 cm−1. δ1H (ppm) o-HC PMePh2 (m) 7.590: m-HC PMePh2 (m) 6.855: p-HC PMePh2 (m) 6.996: HC(Pz) (s) 1H each: 5.726, 5.652, 5.437, H3C(Pz) (s) 3H each: 3.036, 2.470, 2.438, 2.221, 2.172, 2.042: H3C PMePh2 (d) 2.420, 2.402 JHP= 53.399 Hz. δ31P (ppm) 53.392. λmax (dd) 12350 cm−1 (ε, 42 mol−1•cm−1); 22091cm−1 (ε, 128 mol−1•cm−1).
TpMe2MoOCl(OPEtPh2) (2g)
This complex was synthesized following the procedure developed for 2d, with the exception that compound 2 was allowed to react for less than a minute, and produced a golden brown color upon reaction with an excess (two times) of ice cold PEtPh2. Compound 2g was isolated as a yellow-gold powder. νMo=O 947 cm−1. νP=O 1090 cm−1. δ1H (ppm) o-HC OPEtPh2 (dd) 7.724, 7.654: m-HC OPEtPh2 (m) 6.985, 6.906: p-HC OPEtPh2 (m) 7.029: HC(Pz) (s) 1H each: 5.738, 5.662, 5.444, H3C(Pz) (s) 3H each: 3.039, 2.522, 2.423, 2.233, 2.192, 2.054: H2C OPEtPh2 (m) 1.826: H3C OPEtPh2 (dt) 1.144. δ31P (ppm) 57.538. λmax (dd) 12366 cm−1 (ε, 64 mol−1•cm−1); 22030cm−1 (ε, 124 mol−1•cm−1).
TpMe2MoOCl(OPBun3) (2e)
This complex was synthesized following the procedure developed for 2d, with the exception that compound 2 was allowed to react for approximately 1.5 minutes with an excess (two times) of ice cold P(n-Bu)3, and produced a dark green color. Compound 2e was isolated as a dark, olive green powder. δ1H (ppm) HC(Pz) (s) 1H each: 5.795, 5.667, 5.478: H3C(Pz) (s) 3H each: 2.956, 2.921, 2.728, 2.239, 2.184, 2.054: H3C OP(n-Bu)3 9H (t) 0.792: H2C OP(n-Bu)3 (m) 2H 1.867, (m) 2H 1.536, (m) 2H 1.446, (m) 6H 1.349, (m) 6H 1.211. δ31P (ppm) 73.274. νMo=O 953 cm−1. νP=O 1110 cm−1. λmax (dd) 12321 cm−1 (ε, 43 mol−1•cm−1); 22547cm−1 (ε, 147 mol−1•cm−1).
The OAT reactions are described in two steps and the rate constants for the formation of the phosphoryl complex are available; they were treated as a separate process. All rate constants considered in this work were determined under similar reaction conditions, such as temperature, solvent, etc.
Reaction kinetics of TpMoO2X +PR3
The details of kinetic experiments have been reported before,5–8, 18 and similar experimental set ups were used in this investigation. Briefly, solutions of yellow-orange 1, 3, brown 4, and suspensions of 2 were reacted with respective phosphines e.g., PMe3, PEt3, PMe2Ph, PEt2Ph, PBun3, PEtPh2 and PMePh2 producing green to gold solutions containing isolable oxo-Mo(IV)(phosphoryl) complexes, 1a–g, 2a–g, 4a, and 4g while TpMe2MoIVO(OPh)(OPR3) (3a–g) were not isolated due their low stability. The reactions of 1, 3 and 4 follow a second order rate law, dependent on both the Mo-complex and phosphine. All complexes exhibit two low energy presumably dxy dxz/dyz transitions, that are absent in the starting Mo(VI) species. The lowest energy transition is observed ~910 nm (ε 110 M−1cm−1) in the case of TpiPrMoIVO(OPh)(OPEt3) (1c) and TpMe2MoIVO(OPh)(OPEt3) (3c); and at 900 nm (ε, 92M−1 cm−1) in TpiPrMoIVO(OPh)(OPMePh2) (1f) and TpMe2MoIVO(OPh)(OPMePh2) (3f). In complexes 2a–g these transitions are shifted to ~ 800 to 820 nm (ε, 60–90 M−1 cm−1) with the second band appearing as a shoulder or buried beneath the first. In the case of 4a and 4b, these bands are further shifted to 762 nm and 780 nm, respectively. The kinetics for 2 to 2a–2g transformation were not conducted due to the low solubility (solubility < 0.1mg/ml) of 2 in acetonitrile; albeit the same condition was used for 1, 3 and 4. The reaction of 4 with PMe3 yielding 4a has been reported.7 The reactions of 4 with PEtPh2 yielding 4g were conducted in a similar manner, monitoring the reaction at 778 nm. Higher stability of 4a and 4g relative to the corresponding complexes from 1 and 3 allowed for measurements at higher temperatures. The rate constant and activation parameters are tabulated in Supporting Information.
Loss of OPR3 from TpiPrMoO(OPh)(OPR3), TpMe2MoO(OPh)(OPR3) and TpMe2MoO(SPh)(OPR3)
When solutions of 1a–1g, 2a–2g, 3a–3f, or 4a or 4g, are prepared in acetonitrile from the isolated compound or generated in situ, the green to gold colored phosphoryl intermediate forms solvent coordinated complex, 5, 7, or 8, respectively. The details of the kinetics in most cases have been reported, and we used the published data. Transformation of 2a–2g to 6 proceeds from a yellow-green in the case of 2f and 2g, to emerald green or gold in the cases of 2a–2e to a pale blue solution of 6. This reaction is similar to that of the solvolysis of TpiPrMoIVO(OPh)(OPR3), and the variable temperature rate constants are tabulated in the Supporting Information. Similarly, the solvolysis of 4g to 8 was also conducted in acetonitrile and rate constants are tabulated in the Supporting Information.
QALE Analysis
The QALE model has been used as outlined by Giering and Prock,64 as shown in eq 3, where all terms are as defined before. The switching parameter, λ is taken as 0 where θ≤θst, and 1 where θ ≥ θst. The property in question is generally the rate of reaction at 298 K, redox potential, or 31P NMR chemical shift.
First, plots of log k for each reaction were constructed against the QALE phosphorous parameters with special attention to the plot of log k vs θ. The importance of logk vs θ plot, is that a break in the linearity of the plot indicates the presence of a steric threshold, while the lack of apparent breaks suggest that the steric threshold is not operative within the phosphorous species studied. Once the plots were completed and the presence or absence of the steric threshold was determined, the data were examined by multi-component linear regression analysis, with a full set of QALE parameters, using Mini-Tab 14 or a higher version. The phosphine and phosphine oxide parameters are tabulated (Supporting Information). The calculated and observed rates were plotted to determine the quality of the fit. The parameters for individual phosphines were taken from literature values.65–68 However, the phosphine oxide parameters were not easily available; instead of electronic parameters (χd) we used the pKa of phosphine oxides and calculated the cone angles from the crystal structures.69, 70 For phosphoryl species without crystal structures, we approximated cone angles with a similar phosphine oxide e.g., PBun3 with PEt3. The aromatic (Ear) parameters were taken as those of corresponding phosphines. The effect of individual components such as the π acidity parameter and aromatic groups were evaluated by removing that component from the fitting equation and evaluating the quality of fit (eq4–7).
| eq 4 |
| eq 5 |
| eq 6 |
| eq 7 |
The percent contributions of individual parameters to the property in question have been calculated as proposed in the literature.66, 71 For example, the percent contribution of σ-donor ability (% χd) is given by eq 8. Here Δχd, Δθ, ΔEar, and Δπp represent the ranges in the respective parameters.
| eq 8 |
For this analysis, standard values were used, which made a system ligand independent. In this case, the ligand independent nature makes the experiments directly comparable. The parameters used are: Δχd= 40, Δθ= 90, ΔEar = 5 and Δπp= 15. Before applying to an oxygen atom transferring system, the methodology was tested with the rate constants for the oxidation of phosphine with tert-butyl peroxide,72 the results are in excellent agreement with those reported.73
Results
Reaction of [MoVIO2] centers with PR3
The reaction of 4 with PEtPh2 follows a second order kinetics as expected, forming 4g. From variable temperature rate measurements the activation parameters were determined. The reaction follows an associative transition state with negative entropy of activation (ΔS‡= −112 ±17 J • mol−1 K−1). The reaction of 4 with PMe3 was monitored at 726 nm that has a negative entropy of activation (ΔS‡= −172 ±95 J • mol−1 K−1) indicating an associative transition state. While the absolute value is similar to what has been observed before for the reaction with PMe3,7 a large error is associated due to a narrow temperature window and the number of data points.
Compound 2 does not have strong transitions in the visible region, and its reactions with phosphines producing phosphoryl complexes, were not investigated due to the low solubility of the complex under the experimental conditions. Reactions of 3 with PMe3, PMe2Ph, PEt3, PEt2Ph, PBun3, PMePh2, or PEtPh2 follow a bimolecular process with an associative transition state. These reactions are complicated due to the formation of dimeric species which are more prevalent at a higher temperature. Variable temperature rate measurements provided the activation parameters. The negative entropy of activation (−37 to −114 J•mol−1•K−1) is consistent with an associative transition state that has been observed before.
The effect of the ligands on the rate constants for both formation of the phosphoryl complex were examined separately using QALE methodology. In all cases, the rate constants at 298 K were derived from the activation parameters. Steric threshold was included where observed, e.g., the reaction of TpiPrMoO2(OPh) with PR3 exhibited a steric threshold of 128.5° which was included in the analysis (Table 1). However, no steric threshold was observed for TpMe2MoO2(OPh). The multivariable fit are described in the form of equations 9 and 10, respectively for 1 and 3 (Figure 1). The plot for TpMe2MoO2(OPh) indicates a lower R2 and the data are clustered into two domains. We surmise that the analysis of 1 is more reliable.
Table 1.
Rate constants at 298 K for the reaction of TpMoO2(OPh) with PR3 forming TpMoO(OPh)(OPR3).
| TpiPrMoO2(OPh) | TpMe2MoO2(OPh)a | ||
|---|---|---|---|
| Phosphine | logk1b | (Θ-Θst)λ | logk1c |
| PMe3 | −3.058 | 0 | −3.733 |
| PMe2Ph | −2.956 | 0 | −3.605 |
| PEt3 | −3.485 | 3.5 | −3.907 |
| PEt2Ph | −3.555 | 7.5 | 0.658 |
| PBun3 | −3.683 | 7.5 | |
| PMePh2 | −3.830 | 7.5 | 0.508 |
| PEtPh2 | −4.296 | 11.5 | 0.051 |
No steric threshold was detected.
Activation parameters were taken from reference 5, from which the rate constants were calculated.
This work, see the Supporting Information.
Figure 1.
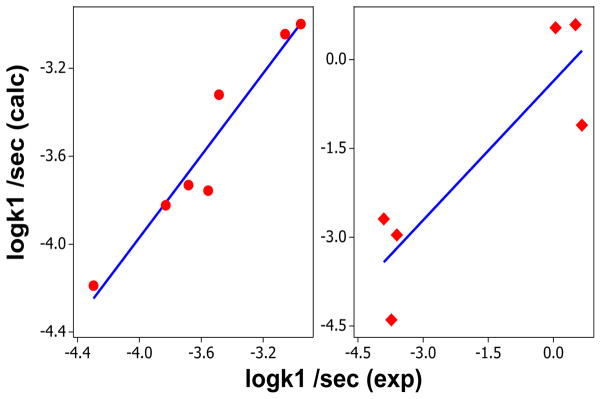
Plot of multicomponent fit of the rate constants (sec−1) at 298 K in acetonitrile with the experimental data. The reactions of phosphines with 1 (circles, left panel) and 3 (diamonds, right panel). The equations 9 and 10 are the fits for compounds 1 and 3, respectively. logk1 = − 2.5 (± 1.3) − 0.063(± 0.16)χd − 0.12 (±0.06)(Θ-Θst)*λ + 0.17 (±0.53) Ear(R2 = 93)… eq 9; logk1 = −34 (± 56) + 0.57 (± 0.57)χd + 0.21 (± 0.34) (Θ-Θst)λ − 0.53 (±5.8) Ear (R2 = 78) … eq10.
From the regression equations, contributions of different factors were calculated. The σ-donor ability (χd) contributes 18 % and 51 % to the reaction of 1 and 3, respectively. Similarly, for the two complexes, the steric factor contributes 76 % and 43 % and the aromatic contribution (Ear) is 6 % for both reactions. However, based on the quality of regression analysis, more confidence is placed on the analysis of 1.
Synthesis of the phosphoryl complexes
Some of the phosphoryl complexes have been reported before and here we disclose the synthesis and characterization of four new phosphoryl complexes (2d–2g). The syntheses of theses complexes followed the same general methodology. The dioxo-MoVI complex, 2, was reacted with a slight excess of the respective phosphine in a nonpolar solvent at a lower temperature. The phosphoryl complexes precipitated from the solution, they were filtered and washed to obtain pure material. The isolated complexes were characterized spectroscopically (UV-visible, IR and NMR) and the spectral data agree well with the previously reported compounds.5, 7–11, 14
QALE analysis of phosphoryl complexes
Using the QALE methodology we have analyzed different properties of the phosphoryl complexes such as the redox potentials, 31P NMR chemical shifts and kinetics of the solvation reactions. The phosphine Ear parameters were used in the analyses. The cone angles were calculated or approximated from the crystal structures following methodology outlined by Tolman21 and Mingos74. For comparison, we conducted the QALE analyses with the phosphine cone angle in the phosphoryl complexes as well. In addition, for the same complexes the pKa values were used instead of electronic parameters as electronic parameters of phosphine oxides are not available.
Analysis of Reduction Potentials
Room temperature cyclic voltammograms of the phosphoryl complexes were recorded in acetonitrile; the voltammograms show an Mo(V/IV) redox couple. In all cases, the TpMe2MoOCl(OPR3) complexes are easier to oxidize than the corresponding TpiPrMoO(OPh)(OPR3), whose potentials were reported previously5 under similar conditions. We attribute this difference is due to two factors; first, the isopropyl groups are more electron donating, which makes the redox potential more negative; and second, the presence of the electron withdrawing chloride group in TpMe2MoOCl(OPR3) makes the oxidation favorable. We surmise that the second factor may be more prominent in modulating the redox potential. For both sets of data, we conducted QALE analyses using the cone angles of phosphine as well as those of phosphine oxide (Table 2). A steric threshold of 135° was detected in TpiPrMoO(OPh)(OPR3) only when the data were analyzed using phosphine cone angles. In general, the R2 values range from 26 % and 31 % in TpiPrMoO(OPh)(OPR3) to 78 % and 75 % in TpMe2MoOCl(OPR3), for phosphine and phosphine oxide angles, respectively. Because of the poor correlations in TpiPrMoO(OPh)(OPR3), we analyzed the TpMe2MoOCl(OPR3) system only (Figure 2). The redox potential can be expressed in terms of equations 11 and 12 for phosphine and phosphine oxide cone angles, respectively. The contribution of each parameter to the redox potential was calculated to be pKa, 7 %;Θ, 38 %, and Ear, 55 % when the phosphine cone angles were used. The corresponding values with phosphine oxide cone angles are pKa, 18 %;Θ, 35 %, and Ear, 47 %; the data suggest the contribution from aromatic factor is more dominant.
Table 2.
Redox potentials and steric parameters used in QALE analyses.
| System | OPR3 | E1/2, mV | (Θ-Θth)λa | (Θ-Θth)λb |
|---|---|---|---|---|
| TpMe2MoOCl(OPR3)d | OPMe3 | −485 | 118 | 115.2 |
| OPMe2Ph | −472 | 136 | 111.7 | |
| OPEt3 | −488 | 132 | 113.5 | |
| OPEt2Ph | −483 | 136 | 111.7c | |
| OPBun3 | −499 | 136 | 113.5 c | |
| OPMePh2 | −472 | 136 | 123 | |
| OPEtPh2 | −468 | 140 | 123 c | |
| TpiPrMoO(OPh)(OPR3)e | OPMe3 | −664 | 0 | 115.2 |
| OPEt3 | −685 | 0 | 113.5 | |
| OPMe2Ph | −688 | 1 | 111.7 | |
| OPPhEt2 | −685 | 1 | 111.7 c | |
| OPBun3 | −700 | 1 | 113.5 c | |
| OPMePh2 | −704 | 1 | 123 | |
| OPEtPh2 | −681 | 5 | 123 c |
Phosphine cone angles were used.
Phosphine oxide cone angles were used.
Approximated.
This work.
Data taken from reference 5.
Figure 2.
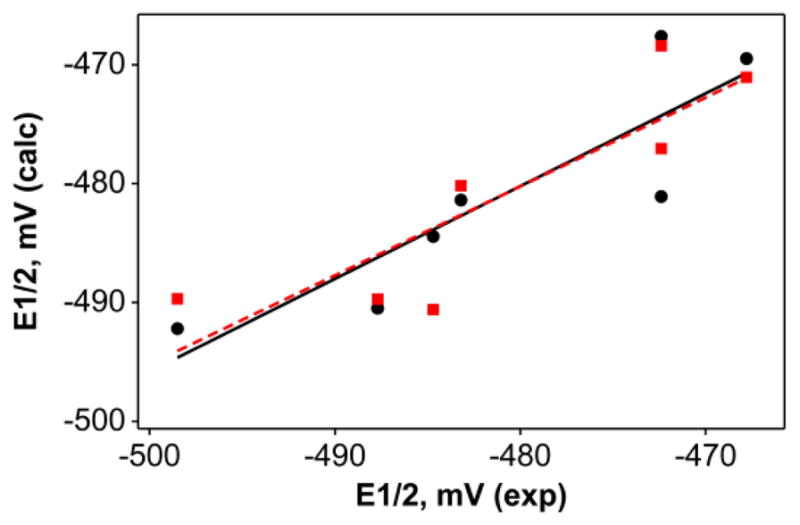
Plot of multicomponent fit of Mo(V/IV) redox potentials (vs Fc+/Fc) of 2a–2g in acetonitrile using phosphine cone angle (circle, eq 11) and phosphine oxide cone angle (square, eq 12): E1/2 = − 434 (± 66.9)+ 0.191 (± 0.97)pKa − 0.431 (±0.52)Θ + 11.4 (±4.17) Ear (R2= 78.0) eq 11; E1/2 = − 432 (± 120) + 0.580 (± 1.13)pKa − 0.520 (±1.07)Θ + 12.4 (±6.62) Ear (R2= 75.9) eq 12
Analysis of 31P NMR Chemical Shift
31P NMR is a good reporter of the coordination of the phosphine oxide to the metal center. Typically, 31P chemical shifts in coordinated phosphine oxides appear downfield compared to the corresponding free phosphine oxide resonance, indicating electron donation from the phosphine oxide to the metal. We analyzed the 31P NMR chemical shift in the phosphoryl complexes with the QALE methodology (Table 3). Again, we used the cone angles of phosphine as well as those of phosphine oxide. We observed steric thresholds of 135.6° and 133.5° in TpMe2MoO(OPh)(OPR3)and TpiPrMoO(OPh)(OPR3), respectively, when the data were analyzed with phosphine cone angles. A steric threshold of 112.4° was observed with phosphine oxide cone angles in TpMe2MoOCl(OPR3). The R2 values for the multi-parameter fit range from 45 % to 97 %, however, R2 for the fits of TpiPrMoO(OPh)(OPR3) are less than 60 %, and were not analyzed further. The equations for the multicomponent fits (Figure 3) for TpMe2MoOCl(OPR3) (eq 13 and 14, using PR3 and OPR3 cone angles) and TpMe2MoO(OPh)(OPR3) (eq 15 and 16, using PR3 and OPR3 cone angles) were used in calculating contributions of different factors to the chemical shifts (Table 4).
Table 3.
31P NMR chemical shift and steric parameters.
| System | OPR3 | δ(31P), ppm | (Θ-Θth)λ a | (Θ-Θth)λ b |
|---|---|---|---|---|
| TpMe2MoOCl(OPR3)d | OPMe3 | 64.1 | 118 | 2.76 |
| OPMe2Ph | 58.0 | 136 | 0 | |
| OPEt3 | 77.4 | 132 | 1.06 | |
| OPEt2Ph | 64.7 | 136 | 0 | |
| OPBun3 | 73.3 | 136 | 1.06 | |
| OPMePh2 | 53.7 | 136 | 10.56 | |
| OPEtPh2 | 57.5 | 140 | 10.56 | |
| TpMe2MoO(OPh)(OPR3)e | OPMe3 | 65 | 0 | 115.2 |
| OPEt3 | 77.9 | 0 | 113.5 | |
| OPEt2Ph | 69.2 | 0.38 | 111.7 c | |
| OPBun3 | 74.6 | 0.38 | 113.5 c | |
| OPEtPh2 | 58.5 | 4.38 | 123 c | |
| TpiPrMoO(OPh)(OPR3)f | OPMe3 | 64 | 0 | 115.2 |
| OPMe2Ph | 58.4 | 2.52 | 111.7 | |
| OPEt3 | 74.9 | 0 | 113.5 | |
| OPEt2Ph | 64.8 | 2.52 | 111.7 c | |
| OPBun3 | 74.4 | 2.52 | 113.5 c | |
| OPMePh2 | 72.8 | 2.52 | 123 | |
| OPEtPh2 | 43.2 | 6.52 | 123 c |
Figure 3.
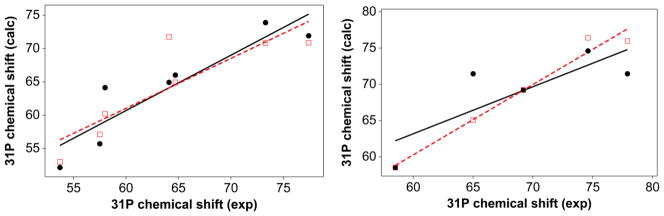
Multicomponent regression analyses of room temperature 31P chemical shifts in benzene. Left panel: 2a–2g, circles, analysis with phosphine cone angles (eq13) and square, analysis with phosphine oxide cone angles (eq 14). Right panel: 3a, 3c–3e, 3g, circles, analysis with phosphine cone angles (eq 15) and square, analysis with phosphine oxide cone angles (eq 16). δ(31P) = 6.30 (±47) −0.39 (± 0.69)pKa+ 0.50 (±0.36) Θ − 10 (±2.9) Ear (R2=83) eq13; δ(31P) = 71 (± 3.3) − 0.78 (± 0.91)pKa + 0.52 (±0.96) (Θ-Θth)λ − 11 (±5.7) Ear (R2=75) eq14; δ(31P) = 74.5 (± 7.15) − 4.20 (± 12.4) pKa + 8.80 (± 31.0) (Θ-Θth)λ − 38.0 (±104) Ear (R2=65) … eq15; δ(31P) = 799 (±210) +8.62 (±2.79)pKa−6.42 (± 1.86) Θ − 51.5 (±17.8) Ear (R2=97) eq 16.
Table 4.
Contributions of different factors to the 31P NMR chemical shift.
| TpMe2MoOCl(OPR3) | TpMe2MoO(OPh)(OPR3) | |||
|---|---|---|---|---|
|
| ||||
| PR3 cone angle | OPR3 cone angle | PR3 cone angle | OPR3 cone angle | |
| pKa | 14 % | 23 % | 15 % | 29 % |
| Cone angle | 40 % | 35 % | 69 % | 49 % |
| Ear | 46 % | 42 % | 16 % | 22 % |
Kinetics of Solvation of Phosphoryl complexes
The solvation of phosphoryl complexes in acetonitrile follows a first order process with the dominant mechanism being dissociative interchange (Id).18 However, in some cases, an associative interchange (Ia) mechanism has been observed.5, 7 The rates of solvation at different temperatures for compounds 1a–1g and 4a have been reported elsewhere.5–8, 18 The solvation reaction of TpMe2MoO(OPh)(OPR3) is complicated due to the formation of dimeric species which precluded obtaining reliable data. Therefore, the kinetics of the solvation reaction of TpMe2MoO(OPh)(OPR3) was not investigated. Solvation reactions of 3a–3g have been investigated in acetonitrile at variable temperatures from which the activation parameters were determined (Table 5). The kinetic data are presented in the supplementary material. In general the reactions follow with a negative entropy of activation, although in TpMe2MoOCl(OPEt2Ph) the value is only −4.1 ± 12 J • mol−1 K−1 indicating that an interchange mechanism is operating. From this value it is difficult to ascertain the nature of the interchange mechanism i.e., associative interchange or dissociative interchange. Solvation of 4g in acetonitrile follows a different trend from 4a and has a positive entropy of activation (39 ± 9.5 J • mol−1 K−1) (Table 5). The variable temperature kinetic data and the activation parameters are placed in the supporting information.
Table 5.
Activation parameters for the solvation of phosphoryl complexes determined in acetonitrile.
| Compound | lnA | Ea (kJ • mol−1) | ΔH‡ (kJ • mol−1) | ΔS‡ (J • mol−1 K−1) | Reference |
|---|---|---|---|---|---|
| TpMe2MoOCl(OPMe3) | 26.8 ± 0.65 | 89.97± 1.6 | 87.46 ± 1.6 | −30.5 ± 5.3 | This work |
| TpMe2MoOCl(OPMe2Ph) | 25.6 ± 2.9 | 84.29± 7.2 | 81.8± 7.2 | −40.4 ± 24.6 | This work |
| TpMe2MoOCl(OPEt3) | 25.0 ± 2.6 | 83.57± 6.3 | 81.13± 6.3 | −44.9 ± 21.6 | This work |
| TpMe2MoOCl(OPEt2Ph) | 29.9 ± 1.4 | 91.77± 3.4 | 89.3 ± 3.4 | −4.1 ± 11.8 | This work |
| TpMe2MoOCl(OPBu3n) | 25.7 ± 2.9 | 84.9± 7.3 | 82.4 ± 7.2 | −39.7 ± 24.5 | This work |
| TpMe2MoOCl(OPMePh2) | 24.0 ± 1.2 | 75.5 ± 3.0 | 73.0 ± 3.4 | −53.8 ± 10.2 | This work |
| TpMe2MoOCl(OPEtPh2) | 23.6 ± 1.0 | 72.5 ± 2.6 | 70.0 ± 2.6 | −57.1 ± 8.7 | This work |
| TpMe2MoO(SPh)(OPEtPh2) | 35.3 ± 1.2 | 99.6 ± 2.9 | 97.0 ± 2.8 | 38.6 ± 9.5 | This work |
| TpMe2MoO(SPh)(OPMe3) | 20.6 ± 0.4 | 70.4 ± 1.4 | 67.8 ± 1.0 | −82.5 ± 3.3 | ref: 7 |
QALE Analysis of Rate Constants of the Solvation Reaction
The room temperature rate constants of the solvation reaction of TpMe2MoOCl(OPR3) and TpiPrMoO(OPh)(OPR3) were analyzed using QALE methodology (Table 6). A steric threshold of 113.0° was observed in TpMe2MoOCl(OPR3) when phosphine oxide cone angles were used. In the case of TpiPrMoO(OPh)(OPR3) the steric threshold is 112.5°. The R2 values of the multivariable fit was in the range from 62 % to 95 %.
Table 6.
QALE parameters for the solvation reaction.
| System | OPR3 | logk2 | (Θ-Θth)λa | (Θ-Θth)λb |
|---|---|---|---|---|
| TpMe2MoOCl(OPR3)d | OPMe3 | −4.13 | 118 | 2.17 |
| OPPhMe2 | −3.66 | 136 | 0 | |
| OPEt3 | −3.77 | 132 | 0.47 | |
| OPEt2Ph | −3.08 | 136 | 0 | |
| OPBun3 | −3.73 | 136 | 0.47 | |
| OPMePh2 | −2.81 | 136 | 9.97 | |
| OPEtPh2 | −2.46 | 140 | 9.97 | |
| TpiPrMoO(OPh)(OPR3)e | OPMe3 | −3.65 | 118 | 2.73 |
| OPMe2Ph | −2.75 | 138 | 0 | |
| OPEt3 | −2.64 | 132 | 1.03 | |
| OPEt2Ph | −2.40 | 136 | 0 | |
| OPMePh2 | −1.94 | 136 | 10.53 | |
| OPEtPh2 | −2.40 | 140 | 10.53 |
Phosphine cone angles were used.
Phosphine oxide cone angles were used.
Approximated.
This work.
Data taken from reference 5.
The multivariable fit, shown in Figure 4, are represented by equations 17 and 18 for TpMe2MoOCl(OPR3) and equations 19 and 20 TpiPrMoO(OPh)(OPR3). These equations were used in calculating the contribution of different factors to the rate of the reactions as tabulated in Table 7.
Figure 4.
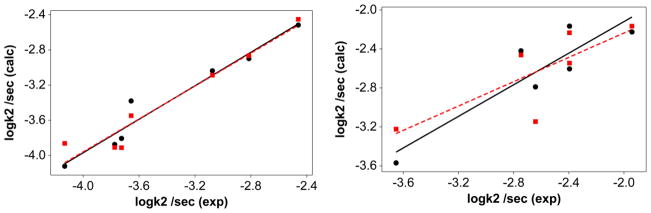
Plot of multicomponent fit of the rate constants at 298 K in acetonitrile for the solvolysis reactions with the experimental data. Left panel: compounds 2a–2g, circles with phosphine cone angles (eq 17) and square with phosphine oxide cone angles (eq 18). logk2 = −6.2 (±1.8) − 0.040 (± 0.26)pKa + 0.18 (±0.01)Θ + 0.38 (±0.11) Ear (R2=95) …eq 17; logk2 = −3.9 (±0.12) − 0.060 (± 0.030)pKa + 0.030(±0.040) (Θ-Θth)λ+0.31 (±0.21) Ear (R2=94)… eq 18.. Right panel: compounds 1a–1g, circles with phosphine cone angles (eq 19) and square with phosphine oxide cone angles (eq 20). logk2 = −10(±4.5) + 0.030(± 0.060)pKa + 0.060(±0.040) Θ+0.180 (±0.270) Ear (R2=81)…eq19; logk2 = −3.1 (±0.35) + 0.020 (± 0.080)pKa − 0.050 (±0.090) (Θ-Θth)λ+0.66 (±0.54) Ear (R2=62) …eq20.
Table 7.
The contribution of different parameters to the rate of solvolysis reaction.
| TpMe2MoOCl(OPR3) | TpiPrMoO(OPh)(OPR3) | |||
|---|---|---|---|---|
|
| ||||
| PR3 cone angle | OPR3 cone angle | PR3 cone angle | OPR3 cone angle | |
| pKa | 32% | 37% | 15% | 10% |
| Cone angle | 31% | 39% | 72% | 50% |
| Ear | 37% | 24% | 13% | 40% |
Discussion
The synthesis of the phosphoryl complexes followed the procedure outlined before.5, 7, 9–11, 14 The reactions of dioxo-Mo(VI) complexes with tertiary phosphines were conducted with slightly excess phosphine and the phosphoryl complexes were precipitated from the reaction solution using very dry solvents and quick processing. The spectroscopic parameters are as expected.
The reaction 4 with PMe3 or PEtPh2 is a second order process that follows associative transition states as evidenced by negative entropy of activation. This reaction is dependent on the nature of the phosphine ligand. In acetonitrile, at 20 °C, PMe3 reacts ~5 times faster than PEtPh2. These two phosphines differ in their steric properties as well as in their basicity. The cone angles are 118° and 140°, respectively for PMe3 and PEtPh2; the corresponding pKas are 8.65 and 4.9. These differences are manifested in a smaller enthalphy of activation, 39.3 kJ mol−1 for PMe3, compared to 56.3 kJ mol−1, for PEtPh2. This difference in enthalpy of activation can also be viewed from its contribution to the free energy of activation. In the case of PMe3 it contributes only 43 %, which increases to 60 % in the case of PEtPh2. A similar behavior has been observed previously in reaction of 1 with PEt3 and PMePh2 which attributed to differences in the basicity and steric properties of the phosphines.18
The reaction of 4 with PMe3 or PEtPh2 leads to formation of the phosphoryl species 4a and 4g, respectively which subsequently solvolyzed to 8. However, the mechanism of solvation changes from associative interchange (Ia) in 4a to dissociative interchange (Id) in 4g as suggested from the entropy of activation (ΔSΘ = −83 ± 3.3 J •mol−1 •K−1 for 4a and 39 ± 9.5 J •mol−1 •K−1 for 4g). We attribute this difference in the mechanism to the steric demand imposed by the leaving group, specifically in the case of OPEtPh2. Because of the difference in the mechanism, we cannot directly compare the two. Regardless of the nature of the transition state, the free energy of activation only varies by 6.0 kJ •mol−1 at 298 K (ΔGΘ, 92 KJ •mol−1 and 86 for 4a and 4g, respectively).
While the rate of phosphoryl complex formation from 3 follows an associative transition state, the entropy of activation varies significantly. At the moment, the origin of this difference is not clear. However, the reaction is still dominated by the enthalpic contribution; ~64–84 % of free energy comes from the enthalpy. There is a large variation (~23 kJ mol−1) in the free energy of activation similar to that in the enthalpy of activation. Unlike in the case of 1, there was no correlation of the rate constant with the cone angle.
The solvolysis of TpMe2MoOCl(OPR3) in acetonitrile follows a second order process. The reaction, however, was monitored under pseudo first order conditions. The reaction proceeds via an associative transition state as indicated by the negative entropy of activation, which varies from −4.1 to −57 J • mol−1 K−1 (Table 5). We suggest that an associative interchange (Id) mechanism may be operating. As mentioned previously, the near zero entropy of activation strongly suggests an interchange mechanism. In this case, enthalpic contribution remains an overwhelmingly dominant factor contributing to 87–97 % of the free energy indicating bond breaking and making in the transition state. Interestingly, the enthalpy of activation varies by 17.5 kJ whereas the free energy varies by 9.50 kJ indicating a slight entropic compensation.
The rate of solvolysis of TpMe2MoOCl(OPR3) is equally dependent on pKa, cone angle, and electronic parameter. In the case of TpiPrMoO(OPh)(OPR3) the cone angle is the is the dominating factor. Between the two ligand systems, the TpiPr is more sterically encumbered at the metal center, therefore expected to exhibit a larger sensitivity towards steric factor. We had previously described an energy controlled pocket created by these tripodal ligands stabilizing the intermediate,9, 10, 12 and the current investigation corroborates the earlier findings. Furthermore, the mechanism of the solvation reaction alters from Id in TpMe2MoOCl(OPR3) to Ia in TpiPrMoO(OPh)(OPR3).
We have also analyzed two physical properties, the reduction potential and 31P NMR chemical shift of the phosphoryl complexes using QALE methodology to understand the ligand effect. Caution must be exercised in interpreting these data due to relatively large errors associated with the parameters. However, in most analyses, the dominant factor is electronic in origin, which includes the pKa and Ear with the steric factor playing a minor but important role. The only difference is in the case of the 31P analysis of TpMe2MoO(OPh)(OPR3) with phosphine cone angles, where the cone angle plays a major role. We suggest that the electronic effect is likely to propagate through the bond and the steric factor controls the orbital interaction which in turn controls the properties. Molecular motion and conformational variations are also to impact the solution properties examined here, which makes it challenging to tease out the exact nature of such interactions, and a detailed computational analysis may help.
The oxygen atom transfer reactivity of TpMe2MoO2(X) (X= Cl, OPh and SPh) with PPh3 has been reported more than 20 years ago, and follows the reactivity order TpMe2MoO2Cl >TpMe2MoO2(SPh) > TpMe2MoO2(OPh).61, 62 Detailed computational analyses, coupled with X-ray absorption studies suggests that the OAT reaction is orbitally controlled and the energy separation between the HOMO and the LUMO of the metal complex is a primary determining factor.75 Determination of the rate constants in a stepwise fashion allows evaluation as to whether the difference in reactivity comes from the electron transfer step or the solvolysis step in the observed overall reaction rates. At room temperature (298 K), in acetonitrile PMe3reacts (first step, k1) with TpMe2MoO2(SPh) 2.5 times faster than TpMe2MoO2(OPh). This is consistent with the general trend of the energies of the frontier orbitals, which parallels redox potentials. In acetonitrile, at 298 K, the solvation (second step, k2) of TpMe2MoO(SPh)(OPMe3) proceeds 5.3 times faster than TpMe2MoOCl(OPMe3). We attribute this difference is due to a stronger Mo—OPMe3 bond in TpMe2MoOCl(OPMe3) as compared to TpMe2MoO(SPh)(OPMe3).
The current investigation attempts to quantitatively assess the major contribution to the reaction rate from the steric factor as represented by cone angles. In both ligand systems, the contribution to the rate is greater than 75%. We have reported previously that the pyrazolyl borate ligands stabilize an energy controlled pocket which helps isolate the phosphoryl complexes, and we anticipate that in a more planar system, the impact may be less. In the case of proteins, where the metal center is strategically placed, it is provocative to suggest that the nucleophilic attack step may be controlled primarily by the steric disposition of the metal center and the incoming substrate.
Summary
The effects of ligands on the oxygen atom transfer reactivity from dioxo-Mo(VI) complexes with tertiary phosphine have been investigated. Under the stated reaction conditions, the reactions are viewed as a two-step process with the first step involving the formation of phosphoryl complexes and the second step as the solvation. The first step follows an associative transition state while the second step can follow either an associative or a dissociative transition state. Quantitative analysis of ligand effect indicates that the first step is dominated by the steric factor, while the electronic factor (sigma donation and aromatic effect) is important. In the second step, other factors such as the pKa and the aromatic effect gain prominence, such that all factors are equally important, especially for TpMe2MoOCl(OPR3). In the case of TpiPrMoO(OPh)(OPR3) the steric factor still dominates. Reaction of PMe3 with TpMe2MoO2(X) (X= Cl, OPh and SPh) indicate only a small difference in the reaction rate which contrasts the reaction with PPh3. The current investigation suggests that even in similarly constituted metal complexes, the extent of ligand’s influence can be different. Thus, it is important to investigate a single system with as many similar substrates as possible to gain detailed insight into the reactivity. A pertinent point in this investigation is that a larger number of data points increases the confidence (e.g., increase in the R2 values), in the QALE analysis, assuming individual data points are associated with low errors. For example, Eyring theory is most commonly used in gaining information about the transition state. In this case, a limited temperature range, necessitated by the instability of the complexes, enhances the errors. The broader aspect of this investigation is that the application of QALE methodology does allow one to tease out the influence of individual components which should be tested further.
Supplementary Material
Acknowledgments
We thank Professor Charles G. Young for stimulating discussions. The project described was supported by Grant Number R15GM061555 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
Supporting Information. Tables of parameters for phosphine, phosphine oxide and variable temperature rate constants.
References
- 1.Hille R. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 2.Tunney JM, McMaster J, Garner CD. Molybdenum and tungsten enzymes. 2004 [Google Scholar]
- 3.Schultz BE, Hille R, Holm RH. J Am Chem Soc. 1995;117:827–828. [Google Scholar]
- 4.Heffron K, Leger C, Rothery RA, Weiner JH, Armstrong FA. Biochemistry. 2001;40:3117–3126. doi: 10.1021/bi002452u. [DOI] [PubMed] [Google Scholar]
- 5.Basu P, Kail BW, Young CG. Inorg Chem. 2010;49:4895–4900. doi: 10.1021/ic902500h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kail BW, Young CG, Johnson ME, Basu P. ACS Symp Ser. 2009;1012:199–217. [Google Scholar]
- 7.Basu P, Nemykin VN, Sengar RS. Inorg Chem. 2009;48:6303–6313. doi: 10.1021/ic900579s. [DOI] [PubMed] [Google Scholar]
- 8.Sengar RS, Nemykin VN, Basu P. J Inorg Biochem. 2008;102:748–756. doi: 10.1016/j.jinorgbio.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nemykin VN, Basu P. Inorg Chem. 2005;44:7494–7502. doi: 10.1021/ic0508324. [DOI] [PubMed] [Google Scholar]
- 10.Millar AJ, Doonan CJ, Smith PD, Nemykin VN, Basu P, Young CG. Chem--Eur J. 2005;11:3255–3267. doi: 10.1002/chem.200401101. [DOI] [PubMed] [Google Scholar]
- 11.Nemykin VN, Laskin J, Basu P. J Am Chem Soc. 2004;126:8604–8605. doi: 10.1021/ja049121f. [DOI] [PubMed] [Google Scholar]
- 12.Nemykin VN, Basu P. Dalton Trans. 2004:1928–1933. doi: 10.1039/b403964e. [DOI] [PubMed] [Google Scholar]
- 13.Nemykin VN, Davie SR, Mondal S, Rubie N, Kirk ML, Somogyi A, Basu P. J Am Chem Soc. 2002;124:756–757. doi: 10.1021/ja017178l. [DOI] [PubMed] [Google Scholar]
- 14.Smith PD, Millar AJ, Young CG, Ghosh A, Basu P. J Am Chem Soc. 2000;122:9298–9299. [Google Scholar]
- 15.Rappe AK, Goddard WA., III J Am Chem Soc. 1980;102:5114–5115. [Google Scholar]
- 16.Rappe AK, Goddard WA., III Nature. 1980;285:311–312. [Google Scholar]
- 17.Pietsch MA, Hall MB. Inorg Chem. 1996;35:1273–1278. doi: 10.1021/ic951044p. [DOI] [PubMed] [Google Scholar]
- 18.Kail BW, Perez LM, Zaric SD, Millar AJ, Young CG, Hall MB, Basu P. Chem--Eur J. 2006;12:7501–7509. doi: 10.1002/chem.200600269. [DOI] [PubMed] [Google Scholar]
- 19.Schuster-Woldan HG, Basolo F. J Am Chem Soc. 1966;88:1657–1663. [Google Scholar]
- 20.Tolman CA. J Am Chem Soc. 1970;92:2956–2965. [Google Scholar]
- 21.Tolman CA. Chem Rev. 1977;77:313–348. [Google Scholar]
- 22.Enemark JH, Cooney JJA, Wang J-J, Holm RH. Chem Rev. 2004;104:1175–1200. doi: 10.1021/cr020609d. [DOI] [PubMed] [Google Scholar]
- 23.Holm RH. Chem Rev. 1987;87:1401–1449. [Google Scholar]
- 24.Holm RH. Coord Chem Rev. 1990;100:183–221. [Google Scholar]
- 25.Holm RH, Solomon EI, Majumdar A, Tenderholt A. Coord Chem Rev. 2011;255:993–1015. [Google Scholar]
- 26.Arzoumanian H. Coord Chem Rev. 1998;178–180:191–202. [Google Scholar]
- 27.Arzoumanian H. Curr Inorg Chem. 2011;1:140–145. [Google Scholar]
- 28.Du G, Abu-Omar MM. Curr Org Chem. 2008;12:1185–1198. [Google Scholar]
- 29.McMaster J, Tunney JM, Garner CD. Prog Inorg Chem. 2003;52:539–583. [Google Scholar]
- 30.Sugimoto H, Tsukube H. Chem Soc Rev. 2008;37:2609–2619. doi: 10.1039/b610235m. [DOI] [PubMed] [Google Scholar]
- 31.Young CG. Biomimetic chemistry of molybdenum. 2000 [Google Scholar]
- 32.Arumuganathan T, Volpe M, Harum B, Wurm D, Belaj F, Mosch-Zanetti NC. Inorg Chem. 2012;51:150–156. doi: 10.1021/ic201308g. [DOI] [PubMed] [Google Scholar]
- 33.Arumuganathan T, Mayilmurugan R, Volpe M, Mosch-Zanetti NC. Dalton Trans. 2011;40:7850–7857. doi: 10.1039/c1dt10248f. [DOI] [PubMed] [Google Scholar]
- 34.Most K, Kopke S, Dall’Antonia F, Mosch-Zanetti NC. Chem Commun (Camb) 2002:1676–1677. doi: 10.1039/b205420e. [DOI] [PubMed] [Google Scholar]
- 35.Das SK, Chaudhury PK, Biswas D, Sarkar S. J Am Chem Soc. 1994;116:9061–9070. [Google Scholar]
- 36.Chaudhury PK, Das SK, Sarkar S. Biochem J. 1996;319:953–959. doi: 10.1042/bj3190953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg JM, Holm RH. J Am Chem Soc. 1984;106:3035–3036. [Google Scholar]
- 38.Bhattacharjee S, Bhattacharyya R. J Chem Soc, Dalton Trans. 1992:1357–1364. [Google Scholar]
- 39.Caradonna JP, Reddy PR, Holm RH. J Am Chem Soc. 1988;110:2139–2144. [Google Scholar]
- 40.Cervilla A, Corma A, Fornes V, Llopis E, Perez F, Rey F, Ribera A. J Am Chem Soc. 1995;117:6781–6782. [Google Scholar]
- 41.Doonan CJ, Slizys DA, Young CG. J Am Chem Soc. 1999;121:6430–6436. [Google Scholar]
- 42.Hammes BS, Chohan BS, Hoffman JT, Einwachter S, Carrano CJ. Inorg Chem. 2004;43:7800–7806. doi: 10.1021/ic049130p. [DOI] [PubMed] [Google Scholar]
- 43.Heinze K, Fischer A. Eur J Inorg Chem. 2010:1939–1947. [Google Scholar]
- 44.Hoffman JT, Einwaechter S, Chohan BS, Basu P, Carrano CJ. Inorg Chem. 2004;43:7573–7575. doi: 10.1021/ic048775m. [DOI] [PubMed] [Google Scholar]
- 45.Keith JM, Tomic ZD, Zaric SD, Hall MB. J Mol Catal A: Chem. 2010;324:15–23. [Google Scholar]
- 46.Laughlin LJ, Young CG. Inorg Chem. 1996;35:1050–1058. doi: 10.1021/ic950815m. [DOI] [PubMed] [Google Scholar]
- 47.Ma X, Schulzke C, Schmidt H-G, Noltemeyer M. Dalton Trans. 2007:1773–1780. doi: 10.1039/b617652f. [DOI] [PubMed] [Google Scholar]
- 48.Marshall-Roth T, Liebscher SC, Rickert K, Seewald NJ, Oliver AG, Brown SN. Chem Commun. 2012;48:7826–7828. doi: 10.1039/c2cc33523a. [DOI] [PubMed] [Google Scholar]
- 49.Over DE, Critchlow SC, Mayer JM. Inorg Chem. 1992;31:4643–4648. [Google Scholar]
- 50.Purohit S, Koley AP, Prasad LS, Manoharan PT, Ghosh S. Inorg Chem. 1989;28:3735–3742. [Google Scholar]
- 51.Roberts SA, Young CG, Cleland WE, Jr, Ortega RB, Enemark JH. Inorg Chem. 1988;27:3044–3051. [Google Scholar]
- 52.Schultz BE, Holm RH. Inorg Chem. 1993;32:4244–4248. [Google Scholar]
- 53.Thiel WR, Eppinger J. Chem--Eur J. 1997;3:696–705. [Google Scholar]
- 54.Tran BL, Carrano CJ. Inorg Chem. 2007;46:5429–5438. doi: 10.1021/ic7005259. [DOI] [PubMed] [Google Scholar]
- 55.Unoura K, Kato Y, Abe K, Iwase A, Ogino H. Bull Chem Soc Jpn. 1991;64:3372–3375. [Google Scholar]
- 56.Volpe M, Moesch-Zanetti NC. Inorg Chem (Washington, DC, U S) 2012;51:1440–1449. doi: 10.1021/ic201681u. [DOI] [PubMed] [Google Scholar]
- 57.Whiteoak CJ, Britovsek GJP, Gibson VC, White AJP. Dalton Trans. 2009:2337–2344. doi: 10.1039/b820754b. [DOI] [PubMed] [Google Scholar]
- 58.Wong Y-L, Ma J-F, Law W-F, Yan Y, Wong W-T, Zhang Z-Y, Mak TCW, Ng DKP. Eur J Inorg Chem. 1999:313–321. [Google Scholar]
- 59.Xiao Z, Bruck MA, Enemark JH, Young CG, Wedd AG. Inorg Chem. 1996;35:7508–7515. doi: 10.1021/ic961983x. [DOI] [PubMed] [Google Scholar]
- 60.Lorber C, Plutino MR, Elding LI, Nordlander E. J Chem Soc, Dalton Trans. 1997:3997–4004. [Google Scholar]
- 61.Roberts SA, Young CG, Kipke CA, Cleland WE, Jr, Yamanouchi K, Carducci MD, Enemark JH. Inorg Chem. 1990;29:3650–3656. [Google Scholar]
- 62.Xiao Z, Bruck MA, Doyle C, Enemark JH, Grittini C, Gable RW, Wedd AG, Young CG. Inorg Chem. 1995;34:5950–5962. doi: 10.1021/ic961983x. [DOI] [PubMed] [Google Scholar]
- 63.Kail B, Nemykin VN, Davie SR, Carrano CJ, Hammes B, Basu P. Inorg Chem. 2002;41:1281–1291. doi: 10.1021/ic011169w. [DOI] [PubMed] [Google Scholar]
- 64.Bartholomew J, Fernandez AL, Lorsbach BA, Wilson MR, Prock A, Giering WP. Organometallics. 1996;15:295–301. [Google Scholar]
- 65.Bunten KA, Chen L, Fernandez AL, Poe AJ. Coord Chem Rev. 2002;233–234:41–51. [Google Scholar]
- 66.Woska D, Prock A, Giering WP. Organometallics. 2000;19:4629–4638. [Google Scholar]
- 67.Golovin MN, Rahman MM, Belmonte JE, Giering WP. Organometallics. 1985;4:1981–1991. [Google Scholar]
- 68.Rahman MM, Liu HY, Prock A, Giering WP. Organometallics. 1987;6:650–658. [Google Scholar]
- 69.Matrosov EI, Cvetkov EI, Mironova ZN, Malevannaay RA, Kabachnik MI. Russian Chemical Bulletin. 1975;24:1231–1234. [Google Scholar]
- 70.Haake P, Cook RD, Hurst GH. J Amer Chem Soc. 1967;89:2650–2653. [Google Scholar]
- 71.Wilson MR, Woska DC, Prock A, Giering WP. Organometallics. 1993;12:1742–1752. [Google Scholar]
- 72.Shulman JI. J Org Chem. 1977;42:3970–3972. [Google Scholar]
- 73.Fernandez AL, Wilson MR, Prock A, Giering WP. Organometallics. 2001;20:3429–3435. [Google Scholar]
- 74.Mueller TE, Mingos DMP. Transition Met Chem. 1995;20:533–539. [Google Scholar]
- 75.Izumi Y, Glaser T, Rose K, McMaster J, Basu P, Enemark JH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1999;121:10035–10046. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.