

Nonsense-mediated decay (NMD) is a key quality control mechanism that targets aberrant mRNAs with premature termination codons (PTCs). Izaurralde and colleagues illustrate the structure of SMG5/SMG7, which are recruited to PTC-containing mRNAs and are required for metazoan NMD. SMG5/SMG7 dimerization is conserved across species and occurs via their 14-3-3 domains in an unusual back-to-back orientation. This study provides further valuable insight into the degradative roles and UPF1-binding affinities of these SMG proteins.
Keywords: mRNA decay, NMD, 14-3-3, UPF1
Abstract
The nonsense-mediated mRNA decay (NMD) pathway triggers the rapid degradation of aberrant mRNAs containing premature translation termination codons (PTCs). In metazoans, NMD requires three 14-3-3-like proteins: SMG5, SMG6, and SMG7. These proteins are recruited to PTC-containing mRNAs through the interaction of their 14-3-3-like domains with phosphorylated UPF1, the central NMD effector. Recruitment of SMG5, SMG6, and SMG7 causes NMD target degradation. In this study, we report the crystal structure of the Caenorhabditis elegans SMG5–SMG7 complex. The 14-3-3-like phosphopeptide recognition domains of SMG5 and SMG7 heterodimerize in an unusual perpendicular back-to-back orientation in which the peptide-binding sites face opposite directions. Structure-based mutants and functional assays indicate that the SMG5–SMG7 interaction is conserved and is crucial for efficient NMD in human cells. Notably, we demonstrate that heterodimerization increases the affinity of the SMG5–SMG7 complex for UPF1. Furthermore, we show that the degradative activity of the SMG5–SMG7 complex resides in SMG7 and that the SMG5–SMG7 complex and SMG6 play partially redundant roles in the degradation of aberrant mRNAs. We propose that the SMG5–SMG7 complex binds to phosphorylated UPF1 with high affinity and recruits decay factors to the mRNA target through SMG7, thus promoting target degradation.
Nonsense-mediated mRNA decay (NMD) is an mRNA quality control mechanism that detects and degrades aberrant mRNAs containing premature translation termination codons (PTCs or nonsense codons) (Rebbapragada and Lykke-Andersen 2009; Nicholson et al. 2010). Therefore, NMD prevents the accumulation of potentially deleterious truncated proteins. In addition to eliminating aberrant mRNAs, NMD post-transcriptionally regulates the expression of naturally occurring transcripts that represent 5%–10% of the eukaryotic transcriptome (Nicholson et al. 2010; Hwang and Maquat 2011).
The molecular mechanism of NMD is best understood in vertebrates, in which a stop codon is efficiently recognized as premature if it is located at least 50–55 nucleotides (nt) upstream of an exon–exon boundary marked by an exon junction complex (EJC) (Le Hir et al. 2000; Nicholson et al. 2010; Hwang and Maquat 2011). Premature translation termination at a PTC leads to the assembly of a “surveillance complex” on the aberrant mRNA that is then degraded (Rebbapragada and Lykke-Andersen 2009; Nicholson et al. 2010).
The conserved core of the surveillance complex consists of the UPF1, UPF2, and UPF3 NMD factors, which are present in all eukaryotes (Rebbapragada and Lykke-Andersen 2009; Nicholson et al. 2010; Hwang and Maquat 2011). In metazoa, additional NMD effectors include the SMG1 kinase, which phosphorylates UPF1, and the SMG5, SMG6, and SMG7 proteins, which in turn trigger UPF1 dephosphorylation (Cali et al. 1999; Yamashita et al. 2001; Anders et al. 2003; Chiu et al. 2003; Ohnishi et al. 2003).
The current NMD models suggest that UPF1 and SMG1 are recruited by ribosomes terminating translation prematurely through interactions with the eRF1 and eRF3 eukaryotic release factors (Czaplinski et al. 1998; Kashima et al. 2006). Upon interaction with UPF2 and/or UPF3 bound to downstream EJCs on an mRNA, UPF1 is phosphorylated by SMG1. In turn, the phosphorylated UPF1 recruits SMG5, SMG6, and SMG7 to the aberrant mRNA (Page et al. 1999; Denning et al. 2001; Yamashita et al. 2001; Anders et al. 2003; Chiu et al. 2003; Ohnishi et al. 2003; Okada-Katsuhata et al. 2011).
SMG5, SMG6, and SMG7 are related proteins that contain an N-terminal domain that consists of nine anti-parallel α helices. The α helices adopt a fold homologous to 14-3-3 proteins, a large family of proteins that bind phosphorylated serines/threonines in their binding partners (Fukuhara et al. 2005; Obsil and Obsilova 2011). Accordingly, SMG5, SMG6, and SMG7 preferentially associate with phosphorylated UPF1 in vivo (Ohnishi et al. 2003; Okada-Katsuhata et al. 2011), and the 14-3-3-like domain of SMG7 binds phosphorylated UPF1 peptides in vitro (Fukuhara et al. 2005). SMG7 has also been shown to interact with SMG5 (Anders et al. 2003; Ohnishi et al. 2003; Unterholzner and Izaurralde 2004; Fukuhara et al. 2005; Okada-Katsuhata et al. 2011).
The SMG5–SMG7 complex and SMG6 have two important functions in NMD: First, they recognize phosphorylated UPF1 and recruit protein phosphatase 2A (PP2A) to promote UPF1 dephosphorylation, which is required for NMD in metazoa (Anders et al. 2003; Chiu et al. 2003; Ohnishi et al. 2003; Franks et al. 2010; Okada-Katsuhata et al. 2011). Second, SMG5, SMG6, and SMG7 are involved in the degradation of PTC-containing mRNAs. SMG6 is a specific NMD endonuclease that cleaves NMD targets in the vicinity of the PTC using its C-terminal PIN (PilT N terminus) domain (Glavan et al. 2006; Huntzinger et al. 2008; Eberle et al. 2009). SMG5 also contains a C-terminal PIN domain, but this domain is inactive (Unterholzner and Izaurralde 2004; Glavan et al. 2006). In contrast, tethered SMG7 promotes the degradation of mRNA reporters (Unterholzner and Izaurralde 2004). This degradative activity is mediated by the SMG7 C-terminal low-complexity region, which is rich in proline residues (PC region) (Fig. 1A; Unterholzner and Izaurralde 2004). SMG7-mediated mRNA degradation involves the decapping enzyme DCP2 and the 5′-to-3′ exonuclease XRN1, suggesting that SMG7 provides a molecular link between the surveillance complex and the general mRNA decay enzymes (Unterholzner and Izaurralde 2004).
Figure 1.
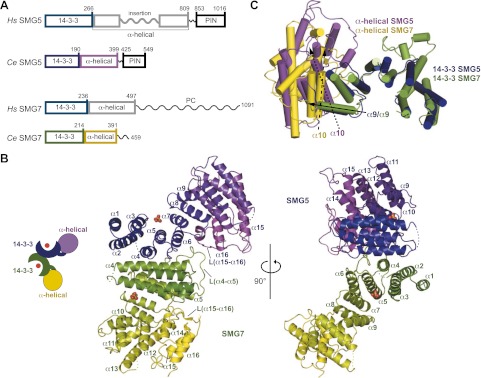
The structure of the Ce SMG5–SMG7 heterodimer. (A) The domain architecture of Hs and Ce SMG5 and SMG7. Hs SMG5 consists of an N-terminal 14-3-3-like domain, a middle α-helical domain containing an insertion of ∼200 amino acids, and a C-terminal inactive PIN domain. Ce SMG5 exhibits a similar domain organization without the long unstructured insertion. Hs SMG7 consists of an N-terminal 14-3-3-like domain, a middle α-helical domain, and a PC region predicted to be unstructured. This domain organization is conserved in Ce SMG7 with the exception of the PC region. The numbers above the protein outlines represent amino acid positions at domain boundaries based on the present structure. (B) Cartoon representation of the overall structure of the Ce SMG5–SMG7 complex. The positions of the phosphate-binding sites of the 14-3-3-like domains are marked by sulfate ions (not included in the final model). Secondary structure elements are indicated. The two views are related by a rotation of 90° around the vertical axis. (C) Comparison of the relative domain orientations in Ce SMG5 and SMG7. The arrows indicate the angle between α helix α9 of the aligned 14-3-3-like domains of SMG5 and SMG7 (filled arrow) relative to α helix α10 of the α-helical domains of SMG5 (dotted arrow) and SMG7 (dashed arrow).
However, a number of questions remain unanswered: What are the relative contributions of SMG6 and the SMG5–SMG7 complex to the degradation of NMD targets? How do SMG5 and SMG7 interact? Is their interaction required for NMD? How do they recognize UPF1?
The presence of N-terminal 14-3-3-like domains in both SMG5 and SMG7 led to a model in which the two proteins heterodimerize using an interface similar to that observed in 14-3-3 proteins. Canonical 14-3-3 proteins homodimerize through the first three α helices of the 14-3-3 domain in a head-to-head orientation, with the phosphopeptide-binding sites of each monomer facing one another in close proximity (Gardino et al. 2006; Obsil and Obsilova 2011). Dimerization is critical for canonical 14-3-3 protein functions and leads to the anchoring of target proteins in close proximity to one another as well as to conformational changes in the target. We therefore asked whether SMG5 and SMG7 would heterodimerize in a manner structurally similar to 14-3-3 dimers and whether target binding would have analogous effects in NMD.
To this end, we determined the crystal structure of the Caenorhabditis elegans (Ce) SMG5–SMG7 heterodimer at a 3.0 Å resolution. We observed that the two proteins interact directly via their 14-3-3-like domains in a previously unobserved mode of interaction. Unlike canonical 14-3-3-like proteins, in the SMG5–SMG7 complex, the two molecules are oriented nearly perpendicular to one another in a back-to-back orientation. Consequently, their phosphate-binding sites face in opposite directions and are spaced much further apart than in canonical 14-3-3 complexes. As a result, the unprecedented arrangement of SMG5 and SMG7 in the complex is suited particularly well to simultaneously bind distant phosphorylated regions of UPF1. Accordingly, we show that the heterodimer binds phosphorylated UPF1 more efficiently than the isolated proteins. The mutagenesis of the residues that are crucial for complex formation indicates that the mode of SMG5–SMG7 interaction is conserved and that the formation of the dimeric complex is functionally important for NMD in human cells.
Results
Structure of the Ce SMG5–SMG7 heterodimer
SMG5 and SMG7 are related proteins that contain two conserved N-terminal domains: a 14-3-3-like domain and an α-helical domain (Fig. 1A; Fukuhara et al. 2005). Relative to Homo sapiens (Hs) SMG7, Hs SMG5 contains an ∼200-residue insertion in its α-helical domain that is predicted to be largely disordered (Fig. 1A).
To gain a detailed understanding of the SMG5–SMG7 interaction, we initially expressed the N-terminal domains of the human proteins in Escherichia coli. However, neither Hs SMG5 nor the Hs SMG5–SMG7 complex could be easily purified, and they formed aggregates in various buffers and were thus unsuitable for crystallization. Sequence analyses of orthologous proteins from other organisms suggested that although Ce SMG5 is highly divergent at the sequence level (18% identity, excluding the insertion), it represents a minimal version of SMG5 without the large disordered insertion (Fig. 1A; Supplemental Fig. S1). Therefore, we chose to determine the structure of the Ce SMG5–SMG7 complex.
Coexpression of the two N-terminal domains of Ce SMG5 and SMG7 in E. coli yielded a heterodimeric protein complex in solution that displayed a favorable biochemical behavior and readily crystallized in space group C2. We solved the structure of the complex by multiwavelength anomalous dispersion and refined it to a 3.0 Å resolution (Supplemental Table S1). The crystals contain two complexes per asymmetric unit. The two SMG5 and SMG7 chains in the asymmetric unit are very similar (overall root mean square deviation [RMSD] of 1.3 Å and 0.7 Å over Cα atoms, respectively), with the exception of some smaller conformational changes in several loops because of different environments in the crystal. The final structural model encompasses residues 2–400 of Ce SMG5 and 1–391 of Ce SMG7, with the exception of a few poorly defined residues in four loops (SMG5 residues 66–67 and 351, and SMG7 residues 76–80 and 279–280). The model was refined with good stereochemistry to a final Rfree of 24.9% (Supplemental Table S1).
Overall structure of the SMG5–SMG7 heterodimers
Sequence alignments indicated that Ce SMG5 and SMG7 contain an N-terminal 14-3-3-like domain; however, the protein sequences downstream from the 14-3-3-like domain exhibited a low identity (<15%) to those of the Hs SMG7 α-helical domain, leading to the suggestion that the α-helical domain was absent in the Ce proteins (Fukuhara et al. 2005). Contrary to expectation, the structure of the Ce SMG5–SMG7 complex revealed that the Ce proteins are structurally similar to Hs SMG7 despite the low sequence identity (Fig. 1B; Supplemental Figs. S1, S2A,B). The N-terminal portions of Ce SMG5 and SMG7 contain two distinct structural domains, as shown for Hs SMG7: an N-terminal 14-3-3-like domain and a C-terminal α-helical domain (Fig. 1B). The 14-3-3-like domain is composed of nine anti-parallel α helices (α1–α9) arranged in helical hairpins. The helices adopt a horseshoe shape wherein the phosphopeptide-binding groove is located on the concave surface (Fig. 1B). The α-helical domain consists of a series of seven anti-parallel α helices (α10–α16) that form a largely globular domain (Fig. 1B).
The structures of Ce SMG5 and SMG7 superpose well to the structure of Hs SMG7 (Fukuhara et al. 2005)—with the RMSD values in the range of 1.9–2.0 Å for the 14-3-3-like domains and 2.0–2.5 Å for the α-helical domains—if the domains are aligned separately (Supplemental Fig. S2A,B). Consequently, Ce SMG5 and SMG7 are also similar to one another (RMSD values of 2.1 Å and 2.4 Å over the Cα atoms for the 14-3-3 and α-helical domains, respectively) (Fig. 1C). However, the orientation of the α-helical domain relative to the 14-3-3-like domain significantly differs between SMG5 and SMG7 (Fig. 1C). This difference is mainly because of the more acute angle of the α-helical domain with respect to the 14-3-3-like domain in SMG7 (∼110° in SMG7 compared with 140° in SMG5).
Both proteins contain extended loops (∼25 residues) connecting the α15 and α16 helices in their respective α-helical domains (Fig. 1B; Supplemental Fig. S2C,D). The loop folds back onto the convex surface of the 14-3-3-like domain, thereby increasing the intramolecular interaction surface of the two domains. A homologous loop is also present in the Hs SMG7 structure (Supplemental Fig. S2E; Fukuhara et al. 2005).
Ce SMG5 closes off the α-helical domain with a long hairpin loop that runs anti-parallel to the last two α helices (Supplemental Fig. S2C). The corresponding loop was deleted from the Ce SMG7 construct, thereby leaving the last two helices accessible for crystal contact. In the Hs SMG7 structure, the equivalent surface was covered by a short β hairpin (Supplemental Fig. S2E; Fukuhara et al. 2005). In addition, Ce SMG7 features a 30-amino-acid-long loop between the α4 and α5 helices [L(α4–α5)] that is prominently involved in the SMG5–SMG7 interaction (Figs. 1B, 2A,C).
Figure 2.
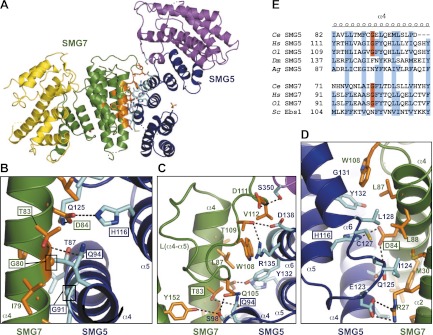
The SMG5–SMG7 interaction interface. (A) An overview of the Ce SMG5–SMG7-binding interface. The residues at the interface between SMG5 and SMG7 are represented as sticks and are highlighted in cyan and orange for SMG5 and SMG7, respectively. (B–D) Enlarged views of the SMG5–SMG7-binding interface. The colors are as in A. The mutated residues in this study are boxed. The views in A and C have similar orientations, and the views in A and D are related by a rotation of ∼180° around the vertical axis. (E) The sequence alignment of α helix α4 of eukaryotic SMG5 and SMG7 orthologs shows the conservation of the glycine residue (highlighted in red) at the center of the binding interface. Conserved hydrophobic residues are highlighted in blue. (Ol) Oryzias latipes (ricefish); (Dm) Drosophila melanogaster; (Ag) Anopheles gambiae; (Sc) Saccharomyces cerevisiae.
The amino acid sequence alignment of the SMG5 and SMG7 14-3-3-like domains from diverse species shows conservation of residues that are distributed throughout the domain (Supplemental Fig. S1). Many of these residues are part of the hydrophobic core of the domain or involved in internal interactions between helices; therefore, these residues are required for the structural integrity of the domain. Furthermore, the 14-3-3-like domains of the two proteins contain conserved residues required for phosphate binding (Y66, R117, and Y118 in SMG5, and K54, R141, and Y142 in SMG7) (Supplemental Figs. S1, S3A,B). We observed positive difference density at the phosphate-binding sites in both monomers, most likely because of sulfate ions from the crystallization condition mimicking phosphate (Supplemental Fig. S3C,D). However, the refinement of sulfate ions in these positions led to unrealistically high B factors for the ligands, most likely because of low occupancies, and they were therefore omitted from the final model. Nevertheless, sulfate ions were placed at these positions in the cartoon diagram representation of the structure to indicate the phosphate-binding site (Fig. 1B; Supplemental Fig. S3). The additional residues lining the phosphopeptide-binding pocket differ between SMG5 and SMG7 (Supplemental Fig. S3A–D), suggesting that they have different specificities for the peptide sequences surrounding the phosphorylated serine/threonine residue.
Heterodimerization interface
SMG5 and SMG7 mainly interact through their 14-3-3-like domains (Figs. 1B, 2A). The dimer buries a total surface area of 2254 Å2 (i.e., ∼1100 Å2 per monomer). The interaction is mediated by an extensive network of hydrogen bonds, salt bridges, π-stacking, and other hydrophobic interactions (Fig. 2A–D). One of the most striking features of the interaction is the perpendicular arrangement of α4 α helices of SMG5 and SMG7 (an angle of ∼88°). This unusual orientation is made possible by the presence of two glycines (SMG5 G91 and SMG7 G80) at the point of contact between the two helices (a distance of 4 Å) (Fig. 2B). These glycine residues allow for the close packing of the two binding partners. Additionally, the residues adjacent to the glycines form intermolecular hydrogen bonds. Specifically, the SMG5 residue Q94 interacts with T83 in SMG7, whereas the SMG5 residues H116 and Q125 make contact with SMG7 D84 (Fig. 2B).
Another important portion of the interaction surface is provided by the loop connecting α helices α4 and α5 of SMG7 [L(α4–α5)] that folds onto the surface of SMG5 (Fig. 2C). In addition to hydrophobic intermolecular interactions, residues in loop L(α4–α5) form several hydrogen bonds with the SMG5 S98, R135, D138, and S350 residues in α helices α4 and α6 and L(α15–α16) and a perpendicular π-stack between W108 (SMG7) and Y132 (SMG5 α helix α6) (Fig. 2C). Notably, the equivalent loop was completely disordered in the structure of the isolated Hs SMG7 monomer (Fukuhara et al. 2005), suggesting that the loop only becomes ordered when binding to SMG5 and may also be involved in the interaction of the human SMG5–SMG7 proteins. More generally, the interacting residues of SMG5 and SMG7 interdigitate similarly to three-dimensional cogs in a wheel and form a dense web of intermolecular bonds (Fig. 2D).
The glycine residue at the center of the interface is well-conserved in SMG5 and SMG7 orthologs (Fig. 2E; Supplemental Fig. S1), with the exceptions of Drosophila melanogaster (Dm) and Anopheles gambiae (Ag) SMG5. This observation is consistent with the absence of an SMG7 ortholog in these insects (Gatfield et al. 2003). Therefore, SMG5 may function as a monomer in these organisms or interact with a functional analog that replaces SMG7 and interacts in a different manner. Conversely, the glycine residue is also absent in Ebs1p, the yeast SMG7 homolog, consistent with the absence of SMG5 homologs in fungi (Luke et al. 2007).
Validation of the interaction interface
We mutated key residues from the dimer interface to test whether Ce SMG5 and Ce SMG7 heterodimerize in solution in a manner similar to that observed in the crystal structure. Specifically, we designed a G91E mutation in SMG5 and a G80E mutation in SMG7. We observed that Ce SMG7 expressed in E. coli with an MBP (maltose-binding protein) tag pulled down Ce SMG5, which was expressed with a GST (glutathione S-transferase) tag (Fig. 3A, lane 5). The interaction was reduced when the pull-down was performed with the Ce SMG7 G80E mutant (Fig. 3A, lane 6). Similarly, the Ce SMG5 G91E mutant was strongly impaired in SMG7 binding (Fig. 3A, lane 7). No interaction was observed between the two mutated proteins (Fig. 3A, lane 8). However, because the interaction of Ce SMG5 with SMG7 is crucial to obtain a soluble recombinant SMG5, the expression levels of GST-Ce SMG5 (wild-type and mutant) were compromised by the coexpression of noninteracting mutants (Fig. 3A, lane 1 vs. lanes 2–4).
Figure 3.
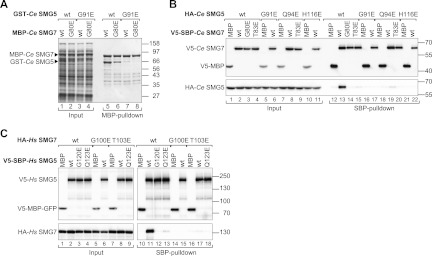
Validation of the interaction interface. (A) GST fusions of Ce SMG5 (wild type or mutants) were coexpressed with MBP fusions of Ce SMG7 (wild type or mutants) in E. coli cells as indicated. MBP-tagged proteins were pulled down using amylose resin. Input samples and bound fractions were analyzed using SDS-PAGE followed by Coomassie blue staining. (B) V5-SBP-tagged Ce SMG7 (wild type or mutants) was coexpressed with HA-tagged Ce SMG5 (wild type or mutants) in HEK293 cells. Ce SMG7 was pulled down from cell lysates using streptavidin Sepharose. V5-SBP-tagged MBP served as a negative control. Inputs (1%) and bound fractions (5% for the V5-tagged proteins and 10% for the HA-tagged proteins) were analyzed by Western blotting. (C) The interaction of HA-tagged Hs SMG7 with V5-SBP-tagged Hs SMG5 (wild type or mutants) was analyzed as described in B. Inputs (1%) and bound fractions (6% for the V5-tagged proteins and 5% for the HA-tagged proteins) were analyzed by Western blotting.
Therefore, we also analyzed the Ce SMG5–SMG7 interaction in coimmunoprecipitation assays in human HEK293 cells, wherein we obtained equal expression levels for all mutants of both proteins. We found that V5-SBP (streptavidin-binding peptide) Ce SMG7 interacted with HA-tagged SMG5 (Fig. 3B, lane 13). The G80E mutation in Ce SMG7 or G91E mutation in Ce SMG5 was sufficient to abrogate the interaction (Fig. 3B, lanes 14, 17). To further probe the interface, we also generated mutations to disrupt central hydrogen bonds. We observed that substitutions of the Ce SMG5 residues Q94 or H116 with glutamic acid were also sufficient to abolish the interaction (Fig. 3B, lanes 19, 22). Conversely, the substitution of Ce SMG7 residue T83 (the binding partner of Q94) with glutamic acid also inhibited complex formation (Fig. 3B, lane 15).
The SMG5–SMG7 interface is conserved
Although the 14-3-3-like domains of the Ce and Hs proteins are not well conserved overall, the key residues involved in heterodimerization are conserved and could be easily mapped onto the human proteins. In particular, the glycine residues in α helix α4 and flanking residues could be unambiguously identified (Supplemental Fig. S1). This similarity enabled us to investigate the effects of mutations of these residues on complex formation in the context of full-length proteins in human cells.
To examine how mutations of glycine residues affect complex formation, V5-SBP-tagged Hs SMG5 was coexpressed with HA-tagged Hs SMG7 in human HEK293 cells. We found that wild-type SMG5 interacted with wild-type SMG7 as previously reported (Ohnishi et al. 2003). The glutamic acid substitution of G120 in Hs SMG5 or G100 in Hs SMG7 (corresponding to G91 and G80 in Ce SMG5 and SMG7, respectively) was sufficient to abrogate the SMG5–SMG7 interaction (Fig. 3C, lanes 12, 15). Similarly, the Q123E mutation in Hs SMG5 (corresponding to Ce SMG5 Q94) strongly reduced complex formation (Fig. 3C, lane 13). Finally, the substitution of the Hs SMG7 residue T103 (corresponding to Ce SMG7 T83) with glutamic acid also abrogated complex formation (Fig. 3C, lane 17). These results suggest that the mode of SMG5–SMG7 interaction is conserved from C. elegans to humans.
The degradative activity of the SMG5–SMG7 complex resides in SMG7
In previous studies, we showed that tethered SMG7 promotes the degradation of an mRNA reporter. This degradation required the PC region (Unterholzner and Izaurralde 2004). In contrast, tethered SMG5 only promoted mRNA decay when coexpressed with SMG7, suggesting that SMG5 has no degradative activity on its own but can promote mRNA degradation by recruiting SMG7. The identification of mutations in SMG5 and SMG7 that abolish their interaction allowed us to investigate the activity of each of these proteins individually in tethering assays without interference from interactions with endogenous SMG5 or SMG7.
We observed that tethered λN-SMG7 caused degradation of a Renilla luciferase (R-Luc) reporter, which contains five Box B hairpins (5BoxB) inserted in its 3′ untranslated region (R-Luc-5BoxB) (Gehring et al. 2003), as was previously shown (Supplemental Fig. S4A,B; Unterholzner and Izaurralde 2004). Similarly, the SMG7 G100E mutant that cannot bind SMG5 also triggered mRNA degradation, indicating that tethered SMG7 causes mRNA degradation independently of SMG5. The activity of SMG7 in tethering assays was also independent of the interaction with UPF1 because mutations in the phosphopeptide-binding site of the 14-3-3-like domain that strongly impairs UPF1 binding (14-3-3 mut, K66E, and R163E) (Fig. 6A,B, below) did not prevent the degradation of the reporter. In contrast to SMG7, tethered λN-SMG5 was inactive (Supplemental Fig. S4A,B). However, λN-SMG5 promoted the degradation of the reporter when coexpressed with SMG7. Furthermore, the SMG5 mutant G120E, which cannot bind SMG7, was inactive in tethering assays regardless of whether it was coexpressed with SMG7 (Supplemental Fig. S4A,B). All proteins were expressed at comparable levels (Supplemental Fig. S4C) and had no inhibitory effects on the expression of an F-Luc reporter without the Box B hairpins (Supplemental Fig. S4D). We conclude that the degradative activity of the SMG5–SMG7 complex resides in SMG7.
Figure 6.

Complex formation between SMG5 and SMG7 is required for high-affinity binding to UPF1. (A) V5-SBP-tagged Hs SMG5 (wild type or mutants) was coexpressed with YFP-tagged Hs SMG7 (wild type or mutants) and HA–tagged Hs UPF1 in HEK293 cells. Hs SMG5 was pulled down from cell lysates using streptavidin Sepharose beads. A V5-SBP-tagged MBP-GFP fusion served as a negative control. Inputs (1%) and bound fractions (5% for the V5 and YFP-tagged proteins and 30% for the HA-tagged proteins) were analyzed by Western blotting. The samples were treated with RNase A before immunoprecipitation. (B) Pull-down of endogenous UPF1 by V5-SBP-tagged Hs SMG5 (wild type or mutants) in the presence of HA-tagged SMG7 (wild type or mutants). Hs SMG5 was pulled down using streptavidin beads. Inputs and bound fractions were analyzed by Western blotting as described in A. (C) Pull-down of endogenous UPF1 by V5-SBP-tagged Hs SMG7 (wild type or mutants) in the presence of wild-type HA-tagged SMG5. (D) Pull-down of endogenous Hs SMG5 and SMG7 by V5-SBP tagged UPF1 (wild type or mutants). (*) A SMG7 degradation product.
SMG5–SMG7 heterodimerization is important for NMD
Next, we used a complementation assay to investigate whether SMG5–SMG7 heterodimerization is important for NMD in human cells. Briefly, shRNAs targeting the ORFs of the smg5 or smg7 mRNAs were used to deplete endogenous SMG5 or SMG7; these depletions inhibit NMD (Gatfield et al. 2003; Luke et al. 2007; Yepiskoposyan et al. 2011). In depleted cells, the SMG5 and SMG7 mutants were then tested for their ability to restore NMD. Transcripts encoding these wild-type and mutant proteins were made resistant to the shRNA with silent mutations.
We monitored NMD using a well-characterized NMD reporter based on the β-globin gene (Thermann et al. 1998). The levels of the PTC-containing β-globin mRNA were reduced ∼30-fold compared with the wild-type levels (data not shown). Depleting SMG5 caused a 2.2-fold increase in the PTC-containing β-globin reporter (Fig. 4A [lane 2], B). In contrast, the expression levels of the corresponding wild-type β-globin mRNA did not change significantly (Fig. 4C,D). A Western blot analysis indicated that the levels of SMG5 in the depleted cells were reduced to <25% of the control levels (Supplemental Fig. S5A). Notably, the levels of endogenous SMG7 were unaffected in SMG5-depleted cells (Supplemental Fig. S5B).
Figure 4.
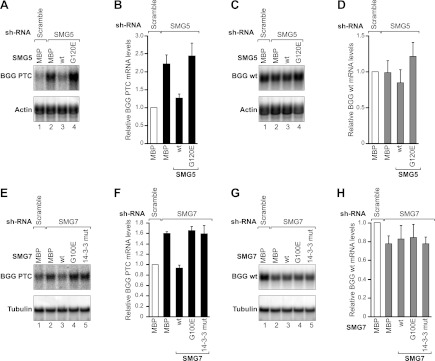
SMG5–SMG7 heterodimerization is required for NMD in human cells. (A–D) HeLa cell lines expressing the β-globin (BGG) NMD reporter (wild type or containing a PTC) were transfected with plasmids expressing the indicated shRNAs. Plasmids expressing an shRNA-resistant version of V5-tagged Hs SMG5 (wild type or mutants) were included in the transfection mixtures, as indicated. V5-MBP served as a negative control. (A,C) The levels of the wild-type or PTC-containing β-globin reporters were analyzed by Northern blotting, normalized to those of the endogenous β-actin mRNA, and set to 1 in control cells (i.e., cells expressing MBP and treated with a scrambled shRNA). B and D show the mean values ± standard deviations obtained in three independent experiments. Note that the Northern blot signals for the wild-type and PTC-containing β-globin reporters cannot be directly compared because the exposure times are not identical. (E–H) A complementation assay with Hs SMG7 was performed as described above for Hs SMG5. (E,G) Northern blot of representative RNA samples. The BGG RNA levels were normalized to endogenous β-tubulin. (F,H) Relative levels of wild-type or PTC-containing β-globin reporters for each condition. The mean values ± standard deviations from three independent experiments are shown.
Expressing the wild-type SMG5 in cells depleted of endogenous protein restored NMD (Fig. 4A [lane 3] B). In contrast, NMD activity was not restored in cells expressing the SMG5 G120 mutant (Fig. 4A [lane 4], B). The wild-type and mutant SMG5 were expressed at comparable levels (Supplemental Fig. S5C).
To further confirm these results, we then performed a complementation assay in SMG7-depleted cells. As observed for SMG5, SMG7 depletion led to a modest increase in the PTC-containing β-globin reporter (Fig. 4E [lane 2], F). The expression levels of the corresponding wild-type β-globin mRNA did not change significantly (Fig. 4G,H). A Western blot analysis indicated that the SMG7 protein levels were reduced to <25% of the control levels in depleted cells, whereas the endogenous SMG5 protein levels were unaffected by the SMG7 depletion (Supplemental Fig. S5D,E).
Similar to the SMG5 observations, wild-type SMG7 restored NMD in SMG7-depleted cells (Fig. 4E [lane 3], F). In contrast, NMD activity was not restored in cells expressing the SMG7 G100E mutant (Fig. 4E, lane 4], F). Similarly, an SMG7 mutant carrying mutations in the 14-3-3-like domain that did not interact with endogenous UPF1 (Fig. 6B, below) also failed to restore NMD (Fig. 4E [lane 5], 4F). The SMG7 mutants were expressed at levels comparable with the wild-type SMG7 (Supplemental Fig. S5F). Collectively, our results indicate that SMG5–SMG7 heterodimerization is required for these proteins to function in NMD.
The SMG5–SMG7 complex acts redundantly with SMG6
Because SMG5 and SMG7 depletion leads to a modest twofold stabilization of the PTC-containing reporter, we investigated whether SMG6 compensates for the absence of the SMG5–SMG7 complex. Two lines of evidence support this possibility. First, SMG6 and SMG7 both trigger the degradation of NMD targets (Gatfield and Izaurralde 2004; Unterholzner and Izaurralde 2004; Huntzinger et al. 2008; Eberle et al. 2009). Second, all three proteins function to promote UPF1 dephosphorylation (Page et al. 1999; Anders et al. 2003; Ohnishi et al. 2003; Okada-Katsuhata et al. 2011).
Therefore, we depleted each protein individually or in combination in human cells. We observed that the depletion of SMG7 led to a 1.6-fold increase in the β-globin-PTC reporter, whereas the depletion of SMG6 resulted in a sixfold increase, as previously reported (Fig. 5A,B; Luke et al. 2007; Kashima et al. 2010). The codepletion of SMG5 or SMG7 in combination with SMG6 resulted in a synergistic inhibition of NMD, leading to a 23-fold or 35-fold increase in the β-globin-PTC mRNA level, respectively (Fig. 5A [lanes 5,6], B). This synergistic effect suggests that SMG6 acts redundantly with the SMG5–SMG7 complex in NMD. Importantly, the codepletion of SMG5 and SMG7 did not exacerbate the effects of the individual SMG5 or SMG7 depletion (a 2.7-fold increase in the reporter mRNA level) (Fig. 5A,B), which is consistent with the idea that SMG5 and SMG7 act together as a complex on the same target mRNA molecule. The codepletions did not affect the expression of the wild-type β-globin reporter (Figs. 5C,D). Western blot analyses indicated that the levels of SMG5 or SMG7 in the depleted cells were reduced to <25% of the control levels (Supplemental Fig. S6A,B).
Figure 5.

The SMG5–SMG7 complex acts redundantly with SMG6. (A–D) HeLa cell lines expressing the β-globin reporter (with or without PTC) were transfected with plasmids expressing the indicated shRNAs. (A,C) The levels of the wild-type or PTC-containing β-globin reporters were analyzed by Northern blot, normalized to those of endogenous β-tubulin mRNA, and set to 1 in control cells (i.e., cells treated with a scrambled shRNA). B and D show the mean values ± standard deviations obtained in three independent experiments. (E–H) A complementation assay with plasmids expressing an siRNA-resistant version of HA-tagged Hs SMG7 (wild type or mutants) was performed in cells codepleted of SMG6 and SMG7. Northern blot analyses of representative RNA samples are shown in E and G. The relative levels of wild-type or PTC-containing β-globin reporters for each condition in three independent experiments are shown in F and H. (I–L) A complementation assay with plasmids expressing shRNA-resistant versions of HA-tagged Hs SMG7 or Hs SMG6 was performed in cells codepleted of endogenous SMG6 and SMG7. RNA samples were analyzed as described above.
Because codepletion of SMG7 and SMG6 provides a broader dynamic range to the complementation assay, we tested the activity of SMG7 mutants in this sensitized background. Figure 5, E and F, shows that the expression of an shRNA-resistant version of wild-type SMG7 partially rescued NMD in cells codepleted of SMG6 and SMG7. In contrast, the SMG7 G100E mutant that did not interact with SMG5 was inactive (Fig. 5E [lane 4], F). Similarly, an SMG7 mutant that binds SMG5 but cannot bind to endogenous phosphorylated UPF1 failed to complement NMD (Fig. 5E [lane 5], F). The levels of the wild-type reporter remained unchanged (Fig. 5G,H). The SMG7 mutants were expressed at levels comparable with the wild-type (Supplemental Fig. S6C). A Western blot analysis revealed that SMG6 and SMG7 codepletion did not affect endogenous SMG5 levels (Supplemental Fig. S6D,E). These results confirm that SMG7 requires interaction with SMG5 and phosphorylated UPF1 to act in the NMD pathway and further indicate that the SMG5–SMG7 complex acts redundantly with SMG6 to promote the degradation of NMD targets.
To further investigate the potential redundancy between SMG6 and SMG7, we tested whether either protein could rescue NMD in cells codepleted of endogenous SMG6 and SMG7. We observed that the expression of either SMG6 or SMG7 partially rescued NMD in codepleted cells and lead to a 1.5-fold reduction in the abundance of the β-globin-PTC mRNA relative to the abundance in cells codepleted of SMG6 and SMG7 expressing MBP (Fig. 5I,J). However, a higher expression level of SMG7 was required to achieve a reduction similar to that observed with SMG6 (Supplemental Fig. S6F), suggesting that SMG6 possesses a greater intrinsic degradative activity. Furthermore, the coexpression of SMG6 and SMG7 had a synergistic effect and rescued NMD more efficiently than the expression of each protein individually (Fig. 5I,J). Remarkably, we observed that when expressed at higher levels, SMG6 alone significantly rescued NMD, leading to a 4.8-fold reduction in the β-globin-PTC mRNA levels compared with the codepleted cells expressing MBP, although we did not observe a full NMD restoration with the maximum amount of expression plasmid that we could transfect in our assay (Supplemental Fig. S6G,H). Nevertheless, the dose-dependent restoration of NMD in codepleted cells by the overexpression of SMG6 suggests that it could completely restore NMD, provided that a higher expression level could be achieved. Alternatively, redundancy may be incomplete. We conclude that SMG6 and SMG7 are partially redundant in promoting the degradation of NMD targets.
The SMG5–SMG7 complex binds UPF1 with higher affinity
Next, we investigated why the heterodimerization of SMG5–SMG7 is important for their function in NMD. Dimerization is critical for the function of canonical 14-3-3 proteins, and target peptides providing two tandem binding motifs have been shown to have an increased affinity for 14-3-3 proteins (Yaffe et al. 1997; Kostelecky et al. 2009). In a similar manner, heterodimerization of SMG5 and SMG7 may serve to arrange their 14-3-3-like binding sites in an optimal orientation for simultaneous binding to two motifs in UPF1. Therefore, we hypothesized that the heterodimer binds UPF1 with higher affinity (because of avidity effects) than isolated monomers. To test this hypothesis, we coexpressed V5-SBP-tagged SMG5 and YFP-SMG7 in human HEK293 cells together with HA-tagged UPF1. Wild-type SMG5 pulled down both SMG7 and UPF1, as expected (Fig. 6A, lane 7). Interestingly, the SMG5 G120E mutant that does not interact with SMG7 exhibited a reduced binding to UPF1 (Fig. 6A, lane 8), suggesting that the SMG5–SMG7 complex binds UPF1 with a higher affinity than the isolated SMG5.
In agreement with the conclusion that SMG5 and SMG7 both contribute to the binding affinity of the complex for UPF1, we observed that the complex formed between wild-type SMG5 and a version of SMG7 carrying mutations in the phosphopeptide-binding site of the 14-3-3-like domain exhibited low UPF1 binding, although SMG5 and SMG7 interacted with one another (Fig. 6A, lane 10).
To validate these results, we also performed pull-downs of endogenous UPF1 with V5-SBP-SMG5 in cells in which HA-SMG7 was coexpressed. We observed that wild-type SMG5 interacted with SMG7 and efficiently pulled down phosphorylated UPF1 (Fig. 6B, lane 6). In contrast, the SMG5 mutant G120E that does not bind SMG7 failed to bind endogenous phosphorylated UPF1 (Fig. 6B, lane 7). Note that in contrast to the pull-down with overexpressed UPF1, the SMG5 G120E mutant showed no residual binding to endogenous UPF1, most likely because the affinity of monomeric SMG5 for endogenous UPF1 is below the detection limit of the assay. In agreement with the results obtained for overexpressed UPF1, the complex formed between wild-type SMG5 and an SMG7 protein carrying mutations in the phosphopeptide-binding site of the 14-3-3-like domain was strongly impaired in binding phosphorylated UPF1, although SMG5 and SMG7 still interacted with one another (Fig. 6B, lane 8).
To ensure that the SMG5–SMG7 complex binds to UPF1 as a complex, we performed the equivalent experiment using V5-SBP-SMG7 as bait. Similarly to the SMG5 pull-down experiment, we only observed a strong binding to phosphorylated UPF1 when SMG7 interacted with SMG5 (Fig. 6C, lane 6). Disruption of SMG7 binding to SMG5 also decreased its binding to phosphorylated UPF1 (Fig. 6C, lane 7). Mutations in the 14-3-3-like domain of SMG7 abolished the binding to UPF1, although this mutant still interacted with SMG5 (Fig. 6C, lane 8). Together, our results indicate that SMG5–SMG7 dimerization is required for high-affinity binding to phosphorylated UPF1. We were not able to test the effects of mutations in the 14-3-3-like domain of SMG5 because mutations of residues involved in phosphopeptide binding (Y88A-R183A or Y88E-R183E) abrogated the binding to SMG7, suggesting that these mutations disrupt the folding of the domain.
It has been reported that SMG5 interacts with the N-terminal region of UPF1 in a phosphorylation-independent manner, whereas the C-terminal phosphorylated Ser1096 is bound by SMG7 as a component of the SMG5–SMG7 complex (Ohnishi et al. 2003; Okada-Katsuhata et al. 2011). Therefore, alanine substitution of Ser1096 is expected to abolish the interaction between UPF1 and the SMG5–SMG7 complex. Accordingly, we observed that wild-type UPF1 coimmunoprecipitated endogenous SMG5 and SMG7, whereas the UPF1 S1096A mutant was strongly impaired in binding to SMG5 and SMG7 (Fig. 6D, lane 8). In contrast, the deletion of the UPF1 N-terminal region had only a minor effect in the binding of the complex (Fig. 6D, lane 9). These results indicate that the peptide surrounding S1096 provides a major binding site for the SMG5–SMG7 complex in UPF1, as reported by Okada-Katsuhata et al. (2011).
Discussion
The structure of the SMG5–SMG7 complex presented in this study shows a 14-3-3-like heterodimer in an unprecedented orientation. This arrangement likely has an impact on the mode of binding to phosphorylated UPF1. The head-to-head dimer formation of canonical 14-3-3 proteins leads the peptides that are bound at the two binding sites to adopt an anti-parallel orientation and brings them into relatively close proximity (a distance of ∼26 Å) (Fig. 7A,B; Rittinger et al. 1999; Obsil and Obsilova 2011). Consequently, the simultaneous binding of two tandem phosphopeptides from one polypeptide chain is only possible if they are spaced at least 10 residues apart (∼32 Å) (Kostelecky et al. 2009). In contrast, in the SMG5–SMG7 heterodimer, the binding sites face away from one another and are tilted relative to one another because of the perpendicular orientation of the 14-3-3-like domains. This domain organization requires a spacer of at least 70 Å between the two target peptides of UPF1, and they would be bound perpendicular to one another (Figs. 7C,D). The structural architecture of the SMG5–SMG7 complex suggests that the binding specificity of the complex for phosphorylated UPF1 arises from three sources: (1) the specificity of SMG5 and SMG7 for different binding sites on UPF1, (2) an avidity effect caused by the heterodimerization of SMG5 and SMG7, and (3) a steric specificity provided by the spatial arrangement of the two 14-3-3-like domains.
Figure 7.

Binding of phosphorylated peptides by the SMG5–SMG7 complex. (A–C) Comparison of the domain orientation of a representative canonical 14-3-3 protein (Protein Data Bank ID: 1qja) (Rittinger et al. 1999) (A) with the SMG5–SMG7 complex (C). Orientation of peptides bound to both binding sites in the 14-3-3 structure (B) and in a model for the SMG5–SMG7 heterodimer (D). The minimal distances are denoted for a peptide linker if both bound peptides are part of one polypeptide chain. (E) Model for the role of the SMG5–SMG7 complex in NMD. The 14-3-3-like domains of the SMG5–SMG7 heterodimer are optimally oriented to bind UPF1 with a high affinity. The question mark indicates that it is unclear whether SMG5 interacts with the N terminus of UPF1 in a phosphorylation-independent manner or with an unknown phosphorylated site in UPF1 or both. SMG7 binds phosphorylated S1096 at the UPF1 C-terminal tail. Both proteins recruit other factors to the target mRNA: The phosphatase PP2A is thought to interact with SMG5, whereas SMG7 recruits the general mRNA decay machinery.
The binding of the SMG5–SMG7 complex to UPF1
The UPF1 protein contains an N-terminal cysteine–histidine (CH) regulatory domain, which provides a binding site for UPF2 (Kadlec et al. 2004; Clerici et al. 2009), and a central helicase domain (Cheng et al. 2007). These structured portions of UPF1 are flanked by the N-terminal and C-terminal unstructured tails (∼100 residues) containing several serine/threonine–glutamine (S/T-Q) motifs that are phosphorylated by SMG1 (Page et al. 1999; Denning et al. 2001; Yamashita et al. 2001; Okada-Katsuhata et al. 2011). Structural studies indicated that UPF1 adopts two main conformations. In the absence of UPF2, UPF1 has a closed conformation in which the N-terminal CH domain interacts with the helicase region (Clerici et al. 2009; Chakrabarti et al. 2011). Upon UPF2 binding, the CH domain undergoes a large conformational change and dissociates from the helicase domain (open conformation), which results in the activation of the UPF1 RNA-unwinding activity (Chamieh et al. 2008; Chakrabarti et al. 2011). Whereas the arrangement of the N-terminal and C-terminal tails in the open and closed conformations is unknown, UPF2 binding is known to be required for SMG1 to phosphorylate UPF1 (Kashima et al. 2006). Therefore, it can be speculated that the N-terminal and C-terminal extensions of UPF1 are only accessible to SMG1 when UPF1 adopts an open conformation as a consequence of UPF2 binding. In this open conformation, the N and C termini are further apart than in the closed conformation. Therefore, the orientation of the 14-3-3-like domains in the SMG5–SMG7 dimer may have evolved to optimally bind the phosphorylated residues in the UPF1 N-terminal and C-terminal tails in only one of the two conformations. In this model, the SMG5–SMG7 heterodimer binding to UPF1 in only a certain conformation would serve as an additional checkpoint to bring about UPF1 dephosphorylation and mRNA degradation by the recruitment of additional factors (Fig. 7E).
Although the above-discussed model is plausible, it is important to note that the exact binding motif for the SMG5 14-3-3-like domain in UPF1 has not been identified. In contrast, it has been shown that SMG7 binds phosphorylated S1096 in the UPF1 C-terminal tail (Okada-Katsuhata et al. 2011). This residue functions as a dominant high-affinity binding site because its presence is required for SMG7 and SMG5 binding to UPF1, indicating that SMG7 tethers the SMG5–SMG7 complex to UPF1 phospho-S1096 (Fig. 6D; Okada-Katsuhata et al. 2011). This observation, together with our finding that mutations in the SMG7 14-3-3-like domain strongly reduce the binding of the SMG5–SMG7 complex to UPF1, indicates that SMG7 provides a major contribution to the binding affinity of the complex for UPF1. This conclusion raises the question regarding the mechanism by which SMG5–SMG7 heterodimerization increases the affinity of the two proteins for UPF1. Analogous to 14-3-3 proteins, we can envisage two possibilities. SMG5 could bind an unphosphorylated motif in the UPF1 N terminus, as previously reported for 14-3-3 proteins (Masters et al. 1999; Ottmann et al. 2007). Alternatively, SMG5 may bind a second phosphorylated low-affinity site on UPF1. Regardless of the precise SMG5-binding site on UPF1, our findings suggest that this site is insufficient on its own for the stable binding of the SMG5–SMG7 complex to UPF1 but is necessary for full binding affinity because of avidity effects. In this scenario, both of the SMG7- and SMG5-binding sites would be required for biological activity. This mode of interaction has also been reported for 14-3-3 proteins (Yaffe 2002). It is important to note that overexpressed SMG5 or SMG7 can interact with overexpressed UPF1 independently of one another (Fig. 6A; Ohnishi et al. 2003; Okada-Katsuhata et al. 2011), which indicates that the isolated SMG5 and SMG7 proteins exhibit some affinity for UPF1. However, it remains unclear whether these interactions contribute to NMD.
The SMG5–SMG7 complex functions redundantly with SMG6 in NMD target degradation
The association of the SMG5–SMG7 complex or SMG6 with phosphorylated UPF1 triggers the degradation of the mRNA target. This degradation occurs through the endonucleolytic cleavage of the mRNA target by the SMG6 PIN domain or recruitment of general mRNA decay enzymes through the SMG7 PC region (Fig. 7E; Nicholson et al. 2010). In this study, we show that these two modes of degradation are partially redundant, with SMG6 providing a major contribution to the degradation of the NMD target analyzed in this study. Two lines of evidence support redundancy between SMG6 and the SMG5–SMG7 complex. First, depletion of SMG5, SMG6, or SMG7 individually had minor inhibitory effects on NMD and resulted in a twofold to sixfold increase in the PTC mRNA levels, whereas the codepletion of SMG6 with either SMG5 or SMG7 had a synergistic effect and resulted in a >20-fold increase in the NMD reporter levels, thus nearly suppressing NMD completely (Fig. 5A). In contrast, the codepletion of SMG5 and SMG7 had no additive effects (Fig. 5A), consistent with the model that these proteins act as a complex. Second, overexpression of SMG6 in cells codepleted of endogenous SMG6 and SMG7 significantly rescued NMD in a dose-dependent manner (Supplemental Fig. S6G,H), indicating that overexpressed SMG6 can compensate for the absence of SMG7 at least partially. However, NMD was not fully restored in this case because of either an inability to express SMG6 at a higher level or an incomplete functional redundancy, as suggested by previous studies (Page et al. 1999; Okada-Katsuhata et al. 2011). Furthermore, our finding that an SMG5 mutant (G91E) that does not interact with SMG7 cannot rescue NMD in cells depleted of endogenous SMG5 demonstrates that SMG5 requires interaction with SMG7 to function in NMD in human cells.
In addition to triggering mRNA target degradation, the association of the SMG5–SMG7 complex or SMG6 with phosphorylated UPF1 elicits a cascade of events, including the recruitment of PP2A, UPF1 dephosphorylation, and recycling of NMD factors to initiate new rounds of NMD (Fig. 7E; Nicholson et al. 2010). Moreover, recent studies indicate that SMG5–SMG7 binding promotes UPF2 dissociation and ribosomal subunit release (Ohnishi et al. 2003; Okada-Katsuhata et al. 2011). The order in which these events occur and the underlying molecular mechanisms remain unknown. Canonical 14-3-3 proteins have been shown to exert their regulatory functions using a variety of mechanisms, including inducing conformational changes in bound proteins, masking binding sites for additional partners, and facilitating interactions between simultaneously bound proteins (Obsil and Obsilova 2011). The SMG5, SMG6, and SMG7 proteins are unique members of the 14-3-3 family because, in addition to the 14-3-3-like domains, they are equipped with additional domains possessing catalytic activity or the ability to bind other proteins. Therefore, the SMG5–SMG7 complex and SMG6 are likely to deploy multiple mechanisms to elicit the events culminating with the destruction of aberrant mRNA and dissociation of the surveillance complex. Understanding the precise molecular mechanism of this NMD effector step remains a challenge for future studies.
Materials and methods
Plasmids, recombinant protein expression, and purification
The plasmids used in this study are described in the Supplemental Material. Ce SMG5 and SMG7 were coexpressed in BL21 star cells (Invitrogen) harboring plasmids encoding GST-Ce SMG5 (1–420) and His6-Ce SMG7 1–395 grown at 37°C in ZY autoinduction medium (Studier 2005) until OD600 = 0.3 was reached. The cell cultures were then incubated overnight at 20°C. After harvest, cells were resuspended in lysis buffer (50 mM HEPES at pH 7.5, 400 mM NaCl, 1 mM DTT, 10% glycerol) supplemented with EDTA-free protease inhibitor (Merck), 1 mg/mL lysozyme, and 5 μg/mL DNase I and lysed by sonication. The cleared lysate was incubated for 1 h with 5 mL of pre-equilibrated glutathione agarose beads (Macherey-Nagel). The beads were washed with lysis buffer, and the protein complex was eluted by incubation with lysis buffer containing 25 mM L-glutathione. Protein tags were cleaved overnight with PreScission protease during dialysis into low-salt buffer (50 mM HEPES at pH 7.5, 200 mM NaCl, 1 mM DTT, 10% glycerol). The complex was subsequently purified over a heparin column followed by a gel filtration column (Superdex 200, GE Healthcare) and stored at −80°C in crystallization buffer (50 mM HEPES at pH 7.5, 300 mM NaCl, 1 mM DTT).
Crystallization and structure determination
Crystals grew at 18°C by hanging-drop vapor diffusion 1 d after mixing 2 μL of protein complex solution (12 mg/mL) with 1 μL of reservoir buffer containing 0.1 M sodium citrate (pH 5.0), 8% (w/v) PEG 8000, 10% glycerol, 500 mM NaCl, 60 mM (NH4)2SO4, 4 mM DTT, and 2 mM TCEP. Before flash-freezing in liquid nitrogen, crystals were soaked in the crystallization buffer with an increased glycerol concentration of 20% for cryoprotection. For phasing, crystals were soaked for 2 h in mother liquor containing 2 mM KAu(CN)2 (Jena Bioscience) and 20% glycerol before flash-freezing. Diffraction of Ce SMG5-SMG7 crystals extended to 3.0 Å for the native and 4.3 Å for the Au-soaked crystals at beamline PXII, Swiss Light Source. Data were recorded on a PILATUS 6M detector (Broennimann et al. 2006) at a wavelength of 0.9793 Å for the native data set. Data sets from two isomorphous Au-soaked crystals were collected at the Au absorption peak at 1.0397 Å and the inflection point at 1.0401 Å. Diffraction images were processed with XDS (Kabsch 2010). Further details on phasing and structure determination are given in the Supplemental Material.
Pull-down assay
MBP-Ce SMG7 1–395 (wild-type or G80E mutant) was coexpressed with GST-Ce SMG5 1–420 (wild-type or G91E mutant) in BL21 star cells in 1 L of ZY autoinduction medium as described above. Cells (6 g) were resuspended in 20 mL of lysis buffer supplemented with 50 mM (NH4)2SO4 and lysed by sonication. After incubation of 7 mL of cleared lysate with 1 mL of amylose resin (New England Biolabs) for 30 min, unbound protein was washed four times with 1 mL of lysis buffer and the bound complexes were eluted twice with 1 mL of lysis buffer containing 25 mM maltose. For analysis, 1.2 μL of lysate and 9 μL of elution were separated on a gradient gel (Any-kD, Bio-Rad).
Coimmunoprecipitation assays and Western blotting
For coimmunoprecipitation assays, HEK293 cells were grown in 10-cm plates and transfected using the calcium phosphate method. To express V5-SBP and HA-tagged proteins, cells were transfected with 24 μg of plasmid. Two days after transfection, cells were lysed for 20 min on ice in RIPA-lysis buffer (20 mM HEPES at pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 2.5 mM MgCl2, 1 mM DTT) supplemented with protease inhibitor (Roche) and 200 μg/mL RNase A (Qiagen). Cell lysates were centrifuged at 16,000g for 15 min at 4°C. The cleared lysate was rotated for 1 h at 4°C with 25 μL of streptavidin Sepharose (GE Healthcare). Beads were washed three times with wash buffer (20 mM HEPES at pH 7.6, 150 mM NaCl, 0.1% NP-40, 2.5 mM MgCl2, 1 mM DTT). Bound proteins were eluted with 100 μL of protein sample buffer and analyzed by Western blotting. For Hs UPF1 coimmunoprecipitations, cells were incubated with 50 nM okadaic acid for 4 h before harvest. NET buffer (50 mM HEPES at pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 2.5 mM MgCl2, 1 mM DTT) supplemented with PhosSTOP phosphatase inhibitor (Roche) was used for lysis and washing. Proteins were detected by Western blotting using the antibodies described in the Supplemental Material.
Tethering and NMD complementation assays
The tethering assay was performed essentially as described before (Lazzaretti et al. 2009). HeLa cell lines stably expressing the β-globin wild-type (BGG wt) or β-globin nonsense codon 39 (BGG PTC) genes were a kind gift from O. Mühlemann (University of Bern) and have been described before (Luke et al. 2007). Plasmids expressing shRNAs for knockdown of NMD factors were derived from the pSUPER plasmid containing the puromycin resistance gene for selection (Yepiskoposyan et al. 2011) and were also supplied by O. Mühlemann. The 19-nt target sequences were as follows: scrambled, ATTCTCCGAACGTGTCACG; SMG5, GAAGGAAATTGGTTGATAC; SMG6, GCTGCAGGTTACTTACAAG; and SMG7, GTATTAGTGCGACACCACT. Cells were grown in DMEM supplemented with 10% heat-inactivated fetal calf serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. HeLa BGG cells were transfected in six-well plates using Lipofectamine 2000 according to the manufacturer's protocol. Transfection mixtures contained 0.4 μg of each sh-plasmid and either 0.2 μg of pV5-SBP-MBP-GFP or 2.4 μg of pV5-SBP-MBP-SMG5. For the SMG7 complementation assays, the transfection mixtures contained either 0.2 μg of pCIneo-λN-HA-MBP or 3 μg of pCIneo-λN-HA-SMG7 with or without 0.8 μg of pCIneo-λN-HA-SMG6. Twenty-four hours after transfection, cells were selected in medium supplemented with 1.5 μg/mL puromycin. After 3.5 d of selection, cells were changed into medium without puromycin for recovery on the evening before cell harvest. Total RNA was isolated using TriFast (Peqlab Biotechnologie) and analyzed as previously described (Unterholzner and Izaurralde 2004). Probes against β-actin or β-tubulin were used as normalization controls.
Accesssion codes
The Protein Data Bank accession code for the structure of the Ce SMG5–SMG7 complex is 3zhe.
Acknowledgments
We are grateful to C. Weiler for technical assistance, A. Boland and R. Büttner for setting up crystallization screens, and J. Braun, E. Huntzinger, and I. Kashima for helpful discussions. We thank the staff at the PX beamlines of the Swiss Light Source for assistance with data collection. This study was supported by the Max Planck Society and by grants from the Deutsche Forschungsgemeinschaft (DFG, FOR855, and the Gottfried Wilhelm Leibniz Program awarded to E.I.) and the European Union Seventh Framework Programme through a Marie Curie Fellowship to S.J. (FP7, n°275343). S.J. is the recipient of an EMBO long-term fellowship.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.206672.112.
References
- Anders KR, Grimson A, Anderson P 2003. SMG-5, required for C. elegans nonsense-mediated mRNA decay, associates with SMG-2 and protein phosphatase 2A. EMBO J 22: 641–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broennimann C, Eikenberry EF, Henrich B, Horisberger R, Huelsen G, Pohl E, Schmitt B, Schulze-Briese C, Suzuki M, Tomizaki T, et al. 2006. The PILATUS 1M detector. J Synchrotron Radiat 13: 120–130 [DOI] [PubMed] [Google Scholar]
- Cali BM, Kuchma SL, Latham J, Anderson P 1999. smg-7 is required for mRNA surveillance in Caenorhabditis elegans. Genetics 151: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Jayachandran U, Bonneau F, Fiorini F, Basquin C, Domcke S, Le Hir H, Conti E 2011. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell 41: 693–703 [DOI] [PubMed] [Google Scholar]
- Chamieh H, Ballut L, Bonneau F, Le Hir H 2008. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol 15: 85–93 [DOI] [PubMed] [Google Scholar]
- Cheng Z, Muhlrad D, Lim MK, Parker R, Song H 2007. Structural and functional insights into the human Upf1 helicase core. EMBO J 26: 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SY, Serin G, Ohara O, Maquat LE 2003. Characterization of human Smg5/7a: A protein with similarities to Caenorhabditis elegans SMG5 and SMG7 that functions in the dephosphorylation of Upf1. RNA 9: 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mourao A, Gutsche I, Gehring NH, Hentze MW, Kulozik A, Kadlec J, Sattler M, Cusack S 2009. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J 28: 2293–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplinski K, Ruiz-Echevarria MJ, Paushkin SV, Han X, Weng Y, Perlick HA, Dietz HC, Ter-Avanesyan MD, Peltz SW 1998. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev 12: 1665–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning G, Jamieson L, Maquat LE, Thompson EA, Fields AP 2001. Cloning of a novel phosphatidylinositol kinase-related kinase: Characterization of the human SMG-1 RNA surveillance protein. J Biol Chem 276: 22709–22714 [DOI] [PubMed] [Google Scholar]
- Eberle AB, Lykke-Andersen S, Mühlemann O, Jensen TH 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55 [DOI] [PubMed] [Google Scholar]
- Franks TM, Singh G, Lykke-Andersen J 2010. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense-mediated mRNA decay. Cell 143: 938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N, Ebert J, Unterholzner L, Lindner D, Izaurralde E, Conti E 2005. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol Cell 17: 537–547 [DOI] [PubMed] [Google Scholar]
- Gardino AK, Smerdon SJ, Yaffe MB 2006. Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin Cancer Biol 16: 173–182 [DOI] [PubMed] [Google Scholar]
- Gatfield D, Izaurralde E 2004. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature 429: 575–578 [DOI] [PubMed] [Google Scholar]
- Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E 2003. Nonsense-mediated mRNA decay in Drosophila: At the intersection of the yeast and mammalian pathways. EMBO J 22: 3960–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE 2003. Y14 and hUpf3b form an NMD-activating complex. Mol Cell 11: 939–949 [DOI] [PubMed] [Google Scholar]
- Glavan F, Behm-Ansmant I, Izaurralde E, Conti E 2006. Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J 25: 5117–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Saulière J, Izaurralde E 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14: 2609–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Maquat LE 2011. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: To die or not to die, that is the question. Curr Opin Genet Dev 21: 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec J, Izaurralde E, Cusack S 2004. The structural basis for the interaction between nonsense-mediated mRNA decay factors UPF2 and UPF3. Nat Struct Mol Biol 11: 330–337 [DOI] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S 2006. Binding of a novel SMG-1–Upf1–eRF1–eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I, Jonas S, Jayachandran U, Buchwald G, Conti E, Lupas AN, Izaurralde E 2010. SMG6 interacts with the exon junction complex via two conserved EJC-binding motifs (EBMs) required for nonsense-mediated mRNA decay. Genes Dev 24: 2440–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostelecky B, Saurin AT, Purkiss A, Parker PJ, McDonald NQ 2009. Recognition of an intra-chain tandem 14-3-3 binding site within PKCɛ. EMBO Rep 10: 983–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaretti D, Tournier I, Izaurralde E 2009. The C-terminal domains of human TNRC6A, TNRC6B, and TNRC6C silence bound transcripts independently of Argonaute proteins. RNA 15: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ 2000. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. EMBO J 19: 6860–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke B, Azzalin CM, Hug N, Deplazes A, Peter M, Lingner J 2007. Saccharomyces cerevisiae Ebs1p is a putative ortholog of human Smg7 and promotes nonsense-mediated mRNA decay. Nucleic Acids Res 35: 7688–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters SC, Pederson KJ, Zhang L, Barbieri JT, Fu H 1999. Interaction of 14-3-3 with a nonphosphorylated protein ligand, exoenzyme S of Pseudomonas aeruginosa. Biochemistry 38: 5216–5221 [DOI] [PubMed] [Google Scholar]
- Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Muhlemann O 2010. Nonsense-mediated mRNA decay in human cells: Mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67: 677–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil T, Obsilova V 2011. Structural basis of 14-3-3 protein functions. Semin Cell Dev Biol 22: 663–672 [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Yamashita A, Kashima I, Schell T, Anders KR, Grimson A, Hachiya T, Hentze MW, Anderson P, Ohno S 2003. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell 12: 1187–1200 [DOI] [PubMed] [Google Scholar]
- Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S 2011. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res 40: 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottmann C, Yasmin L, Weyand M, Veesenmeyer JL, Diaz MH, Palmer RH, Francis MS, Hauser AR, Wittinghofer A, Hallberg B 2007. Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: From structure to pathogenesis. EMBO J 26: 902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page MF, Carr B, Anders KR, Grimson A, Anderson P 1999. SMG-2 is a phosphorylated protein required for mRNA surveillance in Caenorhabditis elegans and related to Upf1p of yeast. Mol Cell Biol 19: 5943–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I, Lykke-Andersen J 2009. Execution of nonsense-mediated mRNA decay: What defines a substrate? Curr Opin Cell Biol 21: 394–402 [DOI] [PubMed] [Google Scholar]
- Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB 1999. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 4: 153–166 [DOI] [PubMed] [Google Scholar]
- Studier FW 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41: 207–234 [DOI] [PubMed] [Google Scholar]
- Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C, Hentze MW, Kulozik AE 1998. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J 17: 3484–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Izaurralde E 2004. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell 16: 587–596 [DOI] [PubMed] [Google Scholar]
- Yaffe MB 2002. How do 14-3-3 proteins work?—Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett 513: 53–57 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC 1997. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S 2001. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev 15: 2215–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Muhlemann O 2011. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 17: 2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]