

Abstract
HIV protease inhibitors (HIV PIs) are the core components of highly active antiretroviral therapies (HAART), which has been successfully used in treatment of HIV-1 infection in the past two decades. However, benefits of HIV PIs are compromised by clinically important adverse effects such as dyslipidemia, insulin resistance and cardiovascular complications. We have previously shown that activation of endoplasmic reticulum (ER) stress plays a critical role in HIV PI-induced dysregulation of hepatic lipid metabolism. HIV PI-induced hepatic lipotoxicity is closely linked to the upregulation of C/EBP homologous protein (CHOP) in hepatocytes. To further investigate whether CHOP is responsible for HIV PI-induced hepatic lipotoxicity, C57BL/6J wild-type (WT) or CHOP knockout (CHOP−/−) mice or the corresponding primary mouse hepatocytes were used in this study. Both in vitro and in vivo studies indicated that HIV PIs (ritonavir and lopinavir) significantly increased hepatic lipid accumulation in WT mice. In contrast, CHOP−/− mice showed a significant reduction in hepatic triglyceride accumulation and liver injury as evidenced by H&E staining and Oil Red O staining. Real-time RT-PCR and immunoblot data showed that in the absence of CHOP, HIV PI-induced expression of stress-related proteins and lipogenic genes was dramatically reduced. Furthermore, TNF-α and IL-6 levels in serum and livers were significantly lower in HIV PI-treated CHOP−/− mice compared to HIV PI-treated WT mice.
CONCLUSION
Taken together, these data suggest that CHOP is an important molecular link of ER stress, inflammation and hepatic lipotoxicity and increased expression of CHOP represents a critical factor underlying events leading to hepatic injury.
Keywords: HAART, ER Stress, Lipid metabolism, CHOP, inflammation
INTRODUCTION
HIV Protease inhibitors (HIV PIs), as the key components of highly active anti-retroviral therapy (HAART), have been successfully used to reduce the morbidity and mortality of HIV-infected patients during last two decades. However, the long-term use of HIV PI therapy is compromised by serious metabolic side effects, such as dyslipidemia, insulin resistance and cardiovascular complications (1–3). Previous studies from our laboratory and other laboratories suggest that HIV PI-induced endoplasmic reticulum (ER) stress response and subsequent activation of the unfolded protein response (UPR) represent important cellular signaling mechanisms of HIV PI-induced metabolic syndromes (4–10).
Emerging evidence indicates that ER stress contributes to the pathogenesis of metabolic and various liver diseases (11–13). The C/EBP homologous protein (CHOP), also known as growth arrest- and DNA damage-inducible gene 153 (GADD153), is a major transcription factor involved in ER stress-mediated apoptosis (14). Recent studies have shown that CHOP is an important regulator of lipotoxicity and oxidative damage in various types of cells (15–20). In addition, our previous studies demonstrated that activation of ER stress and upregulation of CHOP expression are closely linked to HIV PI-induced inflammatory response and foam cell formation in macrophages (5, 7, 8). It also has been reported that CHOP deficiency improved beta cell ultrastructure and promoted cell survival in both genetic and diet-induced mouse models of insulin resistance (19) and ER stress-CHOP-Bax-mediated apoptosis in macrophages contributed to the instability of atherosclerotic plaques (17). However, whether CHOP is the key player in HIV PI-induced hepatic lipotoxicity has not been fully investigated.
The objective of the present study was to examine the role of CHOP in HIV PI-induced dysregulation of hepatic lipid metabolism and further identify the potential underlying mechanisms. The wild type C57BL/6 mice (WT) and CHOP−/− mice as well as the isolated primary mouse hepatocytes were used to determine whether absence of CHOP−/− could prevent HIV PI-induced dyslipidemia and hepatic lipotoxicity. The results indicate that CHOP not only contributed to HIV PI-induced dyslipidemia and hepatic lipid accumulation, but also HIV PI-induced inflammatory response. The identification of a key role of CHOP in ER stress-mediated hepatic lipotoxicity provides novel insights into potential avenues to develop therapeutic strategies for treatment of ER stress-mediated hepatic lipotoxicity associated with drug-induced liver injury and various metabolic diseases.
Materials and methods
Materials
Antibodies against CHOP, activating transcription factor (ATF)-4, X-box-binding protein (XBP)-1, lamin B, horseradish peroxidase-conjugated donkey anti-goat IgG, and horseradish peroxidase-conjugated goat anti-rabbit IgG were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal antibody against β-actin was from Calbiochem (San Diego, CA). Bio-Rad protein assay reagent, Criterion XT Precast Gel, 30% Acrylamide/Bis-acrylamide mix, TEMED and Precision Plus Protein Kaleidoscope standards were obtained from Bio-Rad Laboratories (Hercules, CA). High-capacity cDNA Reverse Transcription kits were from Applied Biosystems (Foster City, CA). SV Total RNA Isolation System was from Promega (Madison, WI). ApoAlert Annexin V kit was from BD Bisociences (San Jose, CA). L-Type TG, H and Cholesterol E kits were from Wako Diagnostics (Richmond, VA). O.C.T gel was from Sakura Finetek (Torrance, CA). Mouse TNF-α and mouse IL-6 ELISA Max™ Set Deluxe Kits were from BioLegend (San Diego, CA). DeadEnd™ Colorimetric TUNEL System was from Progema (Madsion, MI). All other chemical reagents were from Sigma-Aldrich (St. Louis, MO).
Animal studies
C57BL/6J mice, CHOP knockout mice (CHOP−/−, backcrossed at least 12 generations to the C57BL/6J background) were obtained from the Jackson Laboratory (Bar Harbor, ME). All experiments and procedures involving mice were approved by Institutional Animal Care and Use Committee of Virginia Commonwealth University and were conducted in accordance with the Declaration of Helsinki, the Guide for the Care and Use of Animals (National Academies Press, Washington, DC, 1996), and all applicable regulations.
To examine the effect of CHOP on HIV PI-induced lipid accumulation in liver, male WT and CHOP−/− mice (8 weeks old) were randomly assigned to four groups (n=5): 1) control, 2) amprenavir, 3) ritonavir, and 4) lopinavir. Mice were fed with a standard diet and gavaged daily with control solution (0.2% CMC-Na), or individual HIV PIs at a dose of 50 mg/kg for 4 weeks. All mice were housed under identical conditions in an aseptic facility and given free access to water and food. Mice were weighed daily to adjust drug intake. At the end of each time period, mice were fasted for 16 h and blood samples and liver tissues were collected and analyzed.
Isolation and culture of primary mouse hepatocytes
Primary hepatocytes were isolated from C57BL/6 (WT) and CHOP−/− mice (male, 8-week old) and cultured as described previously (6). Cells were cultured in serum-free Williams’ E medium containing dexamethasone (0.1 μM), penicillin (100 units/ml), and thyroxine (1 μM). Cells were incubated from 12 to 24 h in 5% CO2 environment at 37°C before additions were made to culture medium. HIV PIs (Amprenavir, lopinavir, and ritonavir) were dissolved in DMSO and were added directly to culture medium (final concentrations 15 to 50 μM) and incubated for 24 h.
Western blot analysis
The total lysate proteins were prepared from liver tissue using RIPA reagent (20mM Tris-HCl, 150mM NaCl, 2mM EDTA, 20mM NaF, 1mM NaVO4, 1% NP40, and 0.1% SDS, pH 8.0). The protein concentration was determined using Bio-Rad protein assay reagent. The total lysate proteins (100 μg) were resolved on 10% Criterion XT precast gels or 10% SDS-PAGE gel and transferred to nitrocellulose membranes. Immunoreactive bands were detected as described previously (7). The density of the immunoblot bands was analyzed using Bio-Rad Image Lab computer software.
Analysis of apoptosis by annexin V and propidium iodine staining
Mouse primary hepatocytes were treated with HIV PIs (25 μM) for 24 h and stained with Annexin V-FITC and propidium iodide using BD ApoAlert Annexin V kit. Annexin V/propidium iodide-stained cells were visualized under fluorescence microscopy with a 40x objective using a dual-filter set for FITC and rhodamine as described previously (6, 7).
Real-time quantitative PCR
Total cellular RNA was isolated from mouse primary hepatocytes or liver tissues using the SV Total RNA Isolation System. Total RNA (2 μg) was used for the first-strand cDNA synthesis using the High-Capacity cDNA Archive kit. The mRNA levels of key genes involved in hepatic lipid metabolism, such as fatty acid synthase (FAS), cholesterol 7-α-hydroxylase (CYP7A1), sterol 27-hydroxylase (CYP27A1), sterol regulatory element-binding protein-1 (SREBP-1), sterol regulatory element-binding protein-2 (SREBP-2), 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR), and CCAAT enhancer binding protein-beta (C/EBP-β) were quantified using specific primers for each gene (online Table 1). iQ SYBR Green Supermix was used as a fluorescent dye to detect the presence of double-stranded DNA. The mRNA values for each gene were normalized to internal control GAPDH mRNA. The ratio of normalized mean value for each treatment group to vehicle control group was calculated.
Measurement of triglyceride
Mouse primary hepatocytes isolated from WT and CHOP−/− mice were plated on 60-mm plates overnight and then treated with vehicle control or HIV PIs for 24 h. At the end of the treatment, the cells were washed with PBS twice and were harvested with 300 μl RIPA buffer (described in the previous section). Mouse sera were collected at the end of the in vivo study. The liver tissues were homogenized in RIPA buffer. The amount of triglyceride was measured by using the Wako triglyceride assay kit.
Enzyme-linked immunosorbent assays (ELISA) of cytokines
The TNF-α and IL-6 levels in the mouse primary hepatocytes, serum and liver tissue were determined by ELISA using mouse TNF-α and mouse IL-6 ELISA Max™ Set Deluxe Kits as described previously (8). The total protein concentrations of the viable cell pellets and liver tissues were determined using the Bio-Rad Protein Assay reagent. Total amounts of the TNF-α and IL-6 in hepatocytes and liver tissues were normalized to the total protein amounts.
Histopathology analysis
The liver tissue sections were collected and fixed in 4% paraformaldehyde in 0.1 M PBS at room temperature overnight. The regions of the specimens were standardized for all mice. Paraffin-embedded tissue sections (≤ 5μm) were stained with hematoxylin and eosin (H&E) according to standard techniques. The images were taken using a Motic BA200 microscope (Motic Instruments, Inc, Baltimore, MD). Samples were examined in a blindmanner to evaluate the presence of steatosis, inflammation, and fibrosis as described previously (21).
Oil Red O staining
Primary mouse hepatocytes were treated with HIV PIs for 24 h. The intracellular lipid was stained with Oil Red O as described previously (21). The liver tissue sections were collected and covered with O.C.T gel and kept in −80°C. Frozen sections of mouse liver tissue (≤ 10μm) were fixed in 3.7% formaldehyde for 10 min and rinsed with PBS and 60% isopropanol, followed by staining with 0.5% Oil Red O in 60% 2-propanol for 15 min. After washing with distilled water, the nuclei were stained with hematoxylin for 2 min and rinsed thoroughly with distilled water. The images were taken using a microscope equipped with an image recorder under a 40× lens.
TUNEL (TdT-Mediated dUTP Nick-End Labeling) Assay
To detect apoptosis in liver tissue, 5-μm sections were deparaffinized and rehydrated through washes with graded concentrations of ethanol. Tissue was pretreated with proteinase K (20 μg/mL) for 15 minutes at room temperature, followed by incubation in 3% H2O2 in phosphate-buffered saline for 5 minutes at room temperature to quench endogenous peroxidase activity. Apoptotic cells were detected using DeadEnd™ Colorimetric TUNEL System following the manufacturer’s protocol (Promega, Madison, MI). Control stains were obtained by processing, in parallel, duplicate sections omitting only the TnT enzyme.
Statistical analysis
All in vitro experiments were repeated at least three times and results were expressed as the mean ± S.E.M. For in vivo studies, One-way ANOVA analysis of variance was used to analyze the differences between different treatments. Statistics were performed using GraphPad Pro (GraphPad Software Inc., San Diego, CA). A probability (p) of less than 0.05 was considered statistically significant.
Results
Effect of CHOP on HIV PI-induced apoptosis in primary mouse hepatocytes
We have previously reported that HIV PIs induced ER stress and activated the UPR in primary rat hepatocytes. We also showed that HIV PI-induced UPR activation is linked to cell apoptosis and tissue injury (6, 7, 22). Ritonavir and lopinavir are the most commonly used HIV PIs in the clinic and significantly activated the UPR in various cell types including hepatocytes. Similar to our findings in rat primary hepatocytes, both ritonavir and lopinavir dose dependently activated the UPR in primary mouse hepatocytes (online Figure 1). Our previous studies also showed that amprenavir did not induce ER stress nor activate the UPR (6, 7, 22). Therefore, we used amprenavir as a negative control for our studies. In order to determine whether CHOP expression contributed to HIV PI-induced apoptosis in hepatocytes, we isolated primary mouse hepatocytes from wild type and CHOP−/− mice and treated with individual HIV PIs for 24 h. As shown in Fig. 1A, in wild type primary mouse hepatocytes, both ritonavir and lopinavir induced cell apoptosis, but not amprenavir. However, in the CHOP−/− mouse primary hepatocytes, both ritonavir and lopinavir failed to induce apoptosis (Fig. 1B). We also observed that the viability of CHOP−/− mouse primary hepatocytes was higher than that of wild type mouse primary hepatocytes during isolation (data not shown). These results suggest that CHOP may be a key player in HIV PI-induced apoptosis in primary mouse hepatocytes.
Figure 1. Effect of CHOP on HIV PI-induced apoptosis in mouse primary hepatocytes.

Mouse primary hepatocytes (MPH) were isolated from wild type and CHOP−/− mice and treated with vehicle control or individual HIV PIs (25 μM) for 24 h, then stained with Annexin V-FITC and propidium iodide. Images of Annexin V-FITC and propidium iodide-stained cells were visualized under fluorescence microscopy with a dual filter set for FITC and rhodamine. The representative images for each treatment are shown. A. Wild type MPH; B. CHOP−/− MPH. AMPV (amprenavir), RITV (ritonavir) and LOPV (lopinavir).
Effect of CHOP on HIV PI-induced lipid accumulation in primary mouse hepatocytes
We have shown that both ritonavir and lopinavir markedly induced CHOP expression and lipid accumulation in hepatocytes and macrophages (6, 7). To determine whether HIV PIs induce the lipid accumulation through CHOP, we treated both wild type and CHOP−/− mouse primary hepatocytes with individual HIV PIs (15 or 25 μM) for 24 h and the intracellular lipid was stained using Oil Red O as described previously (21). As shown in Fig. 2A, both ritonavir and lopinavir dose-dependently increased intracellular lipid accumulation in wild type mouse primary hepatocytes, but amprenavir had no effect. In contrast, as shown in Fig. 2B, ritonavir and lopinavir-induced intracellular lipid accumulation was significantly reduced in CHOP−/− mouse primary hepatocytes. We further quantified cellular triglyceride (TG) levels. As shown in Fig. 3, in wild type mouse primary hepatocytes, ritonavir and lopinavir dose-dependently increased TG levels, which were markedly inhibited in the absence of CHOP. The basal levels of TG in CHOP−/− mouse primary hepatocytes were much lower than that in wild type mouse primary hepatocytes.
Figure 2. Effect of CHOP on HIV PI-induced lipid accumulation in mouse primary hepatocytes.
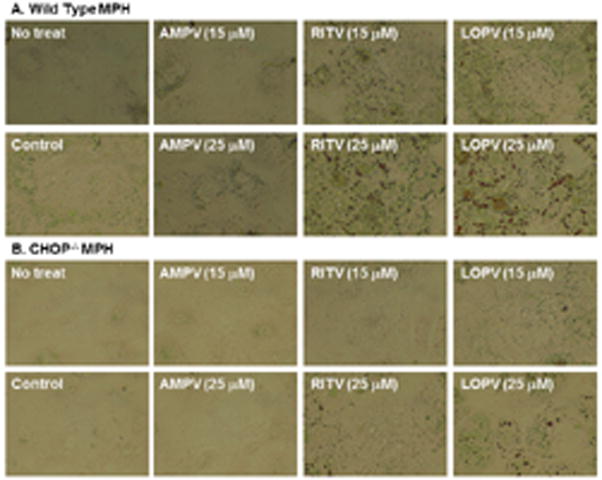
Mouse primary hepatocytes (MPH) were isolated from wild type and CHOP−/− mice and treated with vehicle control or individual HIV PIs (15 or 25 μM) for 24 h. Intracellular lipids were stained with 0.2% Oil Red O as described under Materials and Methods. The images were taken with the use of an Olympus microscope equipped with an image recorder. The representative images for each treatment are shown. AMPV (amprenavir), RITV (ritonavir) and LOPV (lopinavir).
Figure 3. Effect of CHOP on HIV PI-induced increase of triglyceride in mouse primary hepatocytes.

Mouse primary hepatocytes (MPH) were isolated from wild type and CHOP−/− mice and treated with vehicle control or individual HIV PIs (15 or 25 μM) for 24 h. The intracellular triglyceride (TG) levels were measured using Wako TG assay kits according to the protocols provided by the manufacturer. The relative amount of TG was normalized by total protein amount. Values are mean ± SE of three independent experiments. Statistical significance relative to wild type vehicle control, *p < 0.05. **p<0.01 and ***p<0.001. Statistical significance relative to CHOP−/− vehicle control, #p<0.05.
Effect of CHOP on HIV PI-induced dysregulation of the key genes involved in hepatic lipid metabolism in primary mouse hepatocytes
To further identify the cellular mechanisms underlying CHOP-mediated lipid accumulation in hepatocytes, we examined the expression of key genes involved in cholesterol and fatty acid metabolism in HIV PI-treated wild type and CHOP−/− mouse primary hepatocytes by real-time RT-PCR. As shown in Fig. 4, ritonavir and lopinavir-induced increase of SREBP-1, SREBP-2, FAS, HMG-CoAR and C/EBP-β was blunted in CHOP−/− mouse primary hepatocytes. In addition, HIV PI-induced inhibition of CYP7A1, the rate limiting enzyme involved in bile acid synthesis, was reversed in CHOP−/− mouse primary hepatocytes. The Western blot analysis further confirmed that ritonavir- and lopinavir-induced increase of protein expression levels of SREBP1 and SREBP2 in wild type mouse primary hepatocytes was blocked in CHOP−/− mouse primary hepatocytes (Online Figure 2). These results suggest that CHOP contributes to HIV PI-induced increase of cholesterol synthesis and inhibition of bile acid synthesis in hepatocytes.
Figure 4. Effect of CHOP on HIV PI-induced dysregulation of the key genes involved in hepatic lipid metabolism in mouse primary hepatocytes.

The mouse primary hepatocytes (MPH) were isolated from wild type and CHOP−/− mice and treated with vehicle control or individual HIV PIs (25 μM) for 24 h. The total cellular RNA was isolated and reverse transcribed. The relative mRNA levels of SREBP-1, SREBP-2, HMG-CoAR, FAS, CYP7A1, and CEBPβ were determined by real-time PCR as described under Materials and Methods. Values are mean ± SE of three independent experiments; Statistical significance relative to wild type vehicle control, *p < 0.05. **p<0.01 and ***p<0.001.
Effect of CHOP on HIV PI-Induced hepatic lipotoxicity in vivo
To further determine whether CHOP deficiency has a protective effect against HIV PI-induced dysregulation of lipid metabolism and hepatic lipotoxicity in vivo, C57/BL6 wild type and CHOP−/− mice were treated daily with individual HIV PIs for 4 weeks. The serum lipid profile was determined. As shown in Fig. 5A, ritonavir and lopinavir, but not amprenavir, significantly increased serum triglyceride levels in wild type mice, which were diminished in CHOP−/− mice. Similarly, ritonavir and lopinavir induced the increase of triglyceride in the livers of wild type mice, but not in the livers of CHOP−/− mice (Fig 5B). The HE staining and Oil Red O staining results indicated that HIV PI-induced hepatic lipid accumulation was significantly reduced in CHOP−/− mice (Fig. 6A–B). The TUNEL assay further indicated that HIV PI-induced hepatic injury was markedly reduced in CHOP−/− mice (Fig. 6C). The mRNA expression levels of the key genes involved in hepatic lipid metabolism were further analyzed by real-time RT-PCR. As shown in Fig. 7, HIV PI-induced increase of mRNA expression of SREBP-1, SREBP-2, FAS, HMG-CoAR and C/EBP-β was inhibited in CHOP−/− mice. Similarly, the HIV PI-induced down-regulation of CYP7A1 was also reversed in CHOP−/− mice.
Figure 5. Effect of CHOP on HIV PI-induced increase of triglyceride in vivo.

C57BL/6 wild-type and CHOP−/− mice (male, 8 weeks old) were fed with normal diet and gavaged daily with vehicle control (0.2% sodium carboxyl methyl cellulose) or individual HIV PIs (50 mg/kg) for 4 weeks. The TG levels in serum and liver tissue were determined using Wako TG assay kits according to the protocols provided by the manufacturer. The relative amount of TG in liver was normalized by total protein amount. Values are mean ± SE (n=5). Statistical significance relative to wild type vehicle control, *p < 0.05. **p<0.01 and ***p<0.001.
Figure 6. Effect of CHOP on HIV PI-induced hepatic lipid accumulation and injury in vivo.

Wild-type and CHOP−/− mice (male, 8 weeks old) were fed with normal diet and gavaged daily with vehicle control (0.2% sodium carboxy methyl cellulose) or individual HIV PIs (50 mg/kg) for 4 weeks. The liver sections were stained using hematoxylin and eosin (HE) or Oil Red O and the hepatic injury was detected by TUNEL assays as described in “Methods”. The images were taken with an Olympus microscope equipped with an image recorder using a 40x lens. Representative photomicrographs for each treatment are shown. AMPV (amprenavir), RITV (ritonavir) and LOPV (lopinavir). A) HE staining; B) Oil Red O staining; C) TUNEL staining.
Figure 7. Effect of CHOP on HIV PI-induced dysregulation of the key genes involved in hepatic lipid metabolism in vivo.

Wild-type and CHOP−/− mice (male, 8 weeks old) were fed with normal diet and gavaged daily with vehicle control (0.2% sodium carboxy methyl cellulose) or individual HIV PIs (50 mg/kg) for 4 weeks. The total RNA was isolated from liver tissue and reverse transcribed. The relative mRNA levels of SREBP-1, SREBP-2, HMG-CoAR, FAS, CYP7A1, and CEBPβ were determined by real-time PCR as described under Materials and Methods. Values are mean ± SE (n=5); Statistical significance relative to wild type vehicle control, *p < 0.05. **p<0.01 and ***p<0.001.
Effect of CHOP on HIV PI-induced inflammatory response in vivo
It is well-known that inflammation plays a critical role in numerous human diseases including metabolic, cardiovascular and various liver diseases (23–28). ER stress has been identified as an important inducer of inflammation and is closely linked to various inflammatory diseases (29–31). Our previous studies showed that HIV PI-induced ER stress activation contributed to the increase of the expression of inflammatory cytokines, TNF-α and IL-6, in macrophages (5, 8). We also reported that HIV PI-induced ER stress and CHOP expression contributed to the disruption of intestinal epithelial integrity (9). In order to determine whether CHOP expression also induces inflammation response in vivo, we measured TNF-α and IL-6 levels in the serum and liver by ELISA. As shown in Fig. 8, ritonavir and lopinavir, but not amprenavir, significantly increased TNF-α and IL-6 levels in the serum and livers of wild type mice. In contrast, ritonavir and lopinavir failed to increase TNF-α and IL-6 levels in the serum and livers of CHOP−/− mice. These results indicated that CHOP deficiency diminished HIV PI-induced inflammatory response in vivo.
Figure 8. Effect of CHOP on HIV PI-induced inflammation in vivo.

Wild-type and CHOP−/− mice (male, 8 weeks old) were fed with normal diet and gavaged daily with vehicle control (0.2% sodium carboxy methyl cellulose) or individual HIV PIs (50 mg/kg) for 4 weeks. The TNF-α and IL-6 levels in serum and liver tissues were measured by ELISA as described under Materials and Methods. The relative levels of IL-6 and TNF-α in liver tissue were normalized by total protein amounts and expressed as pg/μg protein. Values are mean ± SE (n=5). Statistical significance relative to wild type vehicle control, **p<0.01 and ***p<0.001. Statistical significance relative to CHOP−/− vehicle control, #p<0.05.
Discussion
The highly active antiretroviral therapy (HAART) has been well-documented to markedly reduce the morbidity and mortality of HIV-1 infected patients during the last two decades. However, HAART, especially HIV PI-associated adverse metabolic effects such as dyslipidemia, insulin resistance, and cardiovascular risk have raised major concerns in the clinic (1, 32–35). During last decade, a lot of effort has been put into identifying the potential underlying mechanisms of HIV PI-induced metabolic disorders. Studies from our laboratory and others have identified that activation of ER stress contributes greatly to HIV PI-associated dyslipidemia, hepatic injury and cardiovascular complications (4, 6–10). Our current studies show that CHOP is a key player in HIV PI-induced hepatic lipotoxicity. Both in vitro and in vivo animal studies indicated that knocking out CHOP not only prevented HIV PI-induced dysregulation of lipid metabolism and hepatic injury, but also reduced HIV PI-induced inflammatory response.
The ER is a central organelle of eukaryotic cells which plays a critical role in lipid synthesis, protein folding and protein maturation. Conditions interfering with the function of the ER are collectively called ER stress. The ER has evolved highly-specific signaling pathways termed the Unfolded Protein Response (UPR) to sense cellular stress (36, 37). Recent advances in both basic and clinical studies shed light on mechanistic complexities and on the role of the UPR in numerous diseases including metabolic diseases, cardiovascular diseases, nonalcoholic fatty liver diseases, alcoholic fatty liver diseases, and drug-induced liver diseases (11, 37–44). We have previously shown that HIV PIs induced ER stress and activated the UPR in macrophages, hepatocytes, and intestinal epithelial cells (6, 7, 9). Our studies and studies from others also showed that HIV PIs disrupted lipid metabolism and induced hepatic injury (4, 6, 45). However, the exact linkage between the UPR and HIV PI-mediated hepatic injury remains unclear. CHOP is a major transcription factor involved in ER stress-mediated apoptosis (46–48). Recent studies have shown that CHOP plays multifunctional roles in various diseases and contributes to ER stress-mediated cellular and tissue injury as well as induction of inflammatory response (18, 19, 47–50). Studies done by Ji et al showed that deletion of CHOP remarkably reduced hepatocellular apoptosis in response to alcohol feeding, but had no effect on alcohol-induced hepatic steatosis (18). It has been reported that CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury (49). In addition, CHOP deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes (19). Recent studies further showed that CHOP-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques (17).
In the present study, both ritonavir and lopinavir, the most commonly used HIV PIs in the clinic, dose-dependently activated the UPR, significantly induced apoptosis and increased lipid accumulation in wild type mouse primary hepatocytes, but not in the CHOP−/− mouse primary hepatocytes. Amprenavir, which does not induce activation of the UPR, had no effect on lipid accumulation. Similarly, the in vivo mouse studies showed that ritonavir- and lopinavir-induced increase of serum triglyceride and hepatic lipid accumulation were also blunted in CHOP−/− mice (Fig. 5 and 6). Hepatic steatosis is the result of dysregulation of lipid metabolism caused by increased lipid uptake, increased de novo lipid synthesis, reduced lipid oxidation and metabolism. Further analysis of the effect of HIV PIs on key genes involved in lipid metabolism indicated that CHOP plays an important role in regulating the expression of SREBP-1, SREBP-2, HMG-CoAR, FAS and CYP7A1. In wild type mouse primary hepatocytes and livers, both ritonavir and lopinavir increased the expression of SREBP-1, SREBP-2, HMG-CoAR, FAS, and C/EBPβ, and reduced CYP7A1 expression. However, in the absence of CHOP, ritonavir and lopinavir had no effect on the expression of SREBP-1, SREBP-2, HMG-CoAR, FAS, and C/EBPβ, but increased CYP7A1 expression. Recent studies demonstrated that CHOP knockdown delayed palmitate-mediated cell death in hepatocytes, but had limited effect against methionine-choline-deficient diet (MCD)-induced liver injury (50). Soon et al reported that in a MCD mouse model of fatty liver disease, liver injury is not dependent upon the activation of UPR. CHOP deficiency did not alleviate, and in fact worsened, MCD-mediated liver injury (51). However, deletion of C/EBPβ significantly reduced MCD-induced hepatic triglyceride accumulation and decreased liver injury (52). Our current studies suggest that HIV PI-induced hepatic lipotoxicity is dependent on the UPR activation and CHOP expression. We also noticed that HIV PI-induced c-jun N-terminal Kinases (JNK) activation was partially reduced in the livers of CHOP−/− mice (data not shown). However, the exact molecular/cellular mechanisms underlying CHOP-mediated dysregulation of hepatic lipid metabolism remain to be identified. CHOP may directly regulate the transcription of key genes involved in cholesterol and lipid metabolism or indirectly regulate lipid metabolism through activating other signaling pathways in hepatocytes. Recent advances in the field of autophagy study have identified that the regulation of autophagy and lipid metabolism are interrelated (53, 54). It also has been shown that activation of ER stress is closely linked to autophagy signaling pathway. However, whether HIV PI-induced ER stress and CHOP activation has any effect on hepatic autophagy activity remains to be further studied and is our ongoing project.
Inflammation is an important contributor to hepatic lipotoxicity (25, 28). Our previous studies have reported that HIV PI-induced expression of inflammatory cytokines, TNF-α and IL-6 is linked to ER stress and UPR activation in macrophages (5, 8), and HIV PI-induced CHOP expression plays a role in dysfunction of intestinal barrier (9). In this study, we further demonstrated that HIV PI-induced systemic inflammatory response was also significantly reduced in CHOP−/− mice. Our studies suggest that the ER stress-mediated signaling pathways are emerging as a potential site for the intersection of inflammation and metabolic disease (55).
In summary, together with our previous findings, our studies demonstrated that HIV PI-induced activation of the UPR, especially the upregulation of CHOP represents an important molecular mechanism underlying HIV PI-associated dyslipidemia, inflammation and hepatic lipotoxicity. Our studies suggest that targeting the UPR signaling pathways is a promising and novel approach for reducing HAART-induced metabolic complications in HIV patients. Our studies also provide important information for future development of new anti-HIV drugs with fewer metabolic side effects.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by National Institutes of Health (NIH) Grants R01AT004148 (H.Z), R21AI068432 (H.Z), VA Merit Award 1I01BX001390 (H.Z), The VCU Presidential Research Incentive Program Award (H.Z), Merck Research Grant, School of Medicine, VCU Bridge funding (H.Z), and National Science Foundation of China (Grant 81070245 to H.Z).
We thank the National Institutes of Health AIDS Research and Reference Reagent Program for providing the lopinavir and ritonavir and GlaxoSmithKline for providing amprenavir.
Abbreviations
- HIV PI
HIV protease inhibitor
- HAART
Highly Active Antiretroviral Therapy
- ER
endoplasmic reticulum
- CHOP
C/EBP homologous protein
- UPR
unfolded protein response
- ATF4
activating transcription factor 4
- XBP-1
X-box-binding protein-1
- FAS
fatty acid synthase
- CYP7A1
cholesterol 7-α-hydroxylase
- CYP27A1
sterol 27-hydroxylase
- SREBP-
sterol regulatory element-binding protein-1
- SREBP-2
sterol regulatory element-binding protein-2
- HMG-CoAR
3-hydroxy-3-methyl-glutaryl-CoA reductase
- C/EBP-β
CCAAT enhancer binding protein-beta
- TG
triglyceride
- MCD
methionine-choline-deficient diet
- JNK
c-jun N-terminal Kinase
References
- 1.Palios J, Kadoglou NP, Lampropoulos S. The pathophysiology of HIV-/HAART-related metabolic syndrome leading to cardiovascular disorders: the emerging role of adipokines. Exp Diabetes Res. 2012;2012:103063. doi: 10.1155/2012/103063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wohl DA, McComsey G, Tebas P, Brown TT, Glesby MJ, Reeds D, Shikuma C, et al. Current concepts in the diagnosis and management of metabolic complications of HIV infection and its therapy. Clin Infect Dis. 2006;43:645–653. doi: 10.1086/507333. [DOI] [PubMed] [Google Scholar]
- 3.Kim RJ, Rutstein RM. Impact of Antiretroviral Therapy on Growth, Body Composition and Metabolism in Pediatric HIV Patients. Pediatric Drugs. 2010;12:187–199. doi: 10.2165/11532520-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Parker RA, Flint OP, Mulvey R, Elosua C, Wang F, Fenderson W, Wang S, et al. Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol Pharmacol. 2005;67:1909–1919. doi: 10.1124/mol.104.010165. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Jarujaron S, Wu X, Sun L, Zha W, Liang G, Wang X, et al. HIV protease inhibitor lopinavir-induced TNF-alpha and IL-6 expression is coupled to the unfolded protein response and ERK signaling pathways in macrophages. Biochem Pharmacol. 2009;78:70–77. doi: 10.1016/j.bcp.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou H, Gurley EC, Jarujaron S, Ding H, Fang Y, Xu Z, Pandak WM, Jr, et al. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1071–1080. doi: 10.1152/ajpgi.00182.2006. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H, Pandak WM, Jr, Lyall V, Natarajan R, Hylemon PB. HIV protease inhibitors activate the unfolded protein response in macrophages: implication for atherosclerosis and cardiovascular disease. Mol Pharmacol. 2005;68:690–700. doi: 10.1124/mol.105.012898. [DOI] [PubMed] [Google Scholar]
- 8.Zhou H, Jarujaron S, Gurley EC, Chen L, Ding H, Studer E, Pandak WM, Jr, et al. HIV protease inhibitors increase TNF-alpha and IL-6 expression in macrophages: involvement of the RNA-binding protein HuR. Atherosclerosis. 2007;195:e134–143. doi: 10.1016/j.atherosclerosis.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, Sun L, Zha W, Studer E, Gurley E, Chen L, Wang X, et al. HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Gastroenterology. 2010;138:197–209. doi: 10.1053/j.gastro.2009.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao E, Shinohara M, Feng M, Lau MY, Ji C. Human immunodeficiency virus protease inhibitors modulate Ca(2+) homeostasis and potentiate alcoholic stress and injury in mice and primary mouse and human hepatocytes. Hepatology. 2012 doi: 10.1002/hep.25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozcan L, Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med. 2012;63:317–328. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792–2797. doi: 10.1161/ATVBAHA.111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108:2777–2793. doi: 10.1002/bit.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dasari B, Prasanthi JR, Marwarha G, Singh BB, Ghribi O. The oxysterol 27-hydroxycholesterol increases beta-amyloid and oxidative stress in retinal pigment epithelial cells. BMC Ophthalmol. 2010;10:22. doi: 10.1186/1471-2415-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasanthi JR, Larson T, Schommer J, Ghribi O. Silencing GADD153/CHOP gene expression protects against Alzheimer’s disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS One. 2011;6:e26420. doi: 10.1371/journal.pone.0026420. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, Yano M, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–1932. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 18.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem. 2012;151:217–219. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Zhang L, Gurley E, Studer E, Shang J, Wang T, Wang C, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47:1905–1915. doi: 10.1002/hep.22239. [DOI] [PubMed] [Google Scholar]
- 22.Zhou H. HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Methods Enzymol. 2010;490:107–119. doi: 10.1016/B978-0-12-385114-7.00006-4. [DOI] [PubMed] [Google Scholar]
- 23.Rath E, Haller D. Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur J Nutr. 2011;50:219–233. doi: 10.1007/s00394-011-0197-0. [DOI] [PubMed] [Google Scholar]
- 24.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15:635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navab M, Gharavi N, Watson AD. Inflammation and metabolic disorders. Curr Opin Clin Nutr Metab Care. 2008;11:459–464. doi: 10.1097/MCO.0b013e32830460c2. [DOI] [PubMed] [Google Scholar]
- 26.Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in Alcoholic Liver Disease. Annu Rev Nutr. 2012 doi: 10.1146/annurev-nutr-072610-145138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2012;74:213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- 28.Harmon RC, Tiniakos DG, Argo CK. Inflammation in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2012;5:189–200. doi: 10.1586/egh.11.21. [DOI] [PubMed] [Google Scholar]
- 29.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- 30.Kaser A, Blumberg RS. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 2010;3:11–16. doi: 10.1038/mi.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gotoh T, Endo M, Oike Y. Endoplasmic reticulum stress-related inflammation and cardiovascular diseases. Int J Inflam. 2011;2011:259462. doi: 10.4061/2011/259462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bongiovanni M, Casana M, Tincati C, d’Arminio Monforte A. Treatment interruptions in HIV-infected subjects. J Antimicrob Chemother. 2006;58:502–505. doi: 10.1093/jac/dkl268. [DOI] [PubMed] [Google Scholar]
- 33.Bongiovanni M, Bini T, Capetti A, Trovati S, Di Biagio A, Tordato F, Monforte A. Long-term antiretroviral efficacy and safety of lopinavir/ritonavir in HAART-experienced subjects: 4 year follow-up study. Aids. 2005;19:1934–1936. doi: 10.1097/01.aids.0000189569.10471.b4. [DOI] [PubMed] [Google Scholar]
- 34.Bongiovanni M, Bini T, Casana M, Cicconi P, Tordato F, Monforte AD. Long-term immunologic outcome in HAART-experienced subjects receiving lopinavir/ritonavir. AIDS Res Hum Retroviruses. 2006;22:1096–1105. doi: 10.1089/aid.2006.22.1096. [DOI] [PubMed] [Google Scholar]
- 35.Meraviglia P, Schiavini M, Castagna A, Vigano P, Bini T, Landonio S, Danise A, et al. Lopinavir/ritonavir treatment in HIV antiretroviral-experienced patients: evaluation of risk factors for liver enzyme elevation. HIV Med. 2004;5:334–343. doi: 10.1111/j.1468-1293.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- 36.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 37.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 38.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Flamment M, Kammoun HL, Hainault I, Ferre P, Foufelle F. Endoplasmic reticulum stress: a new actor in the development of hepatic steatosis. Curr Opin Lipidol. 2010;21:239–246. doi: 10.1097/MOL.0b013e3283395e5c. [DOI] [PubMed] [Google Scholar]
- 41.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 42.Nagy G, Kardon T, Wunderlich L, Szarka A, Kiss A, Schaff Z, Banhegyi G, et al. Acetaminophen induces ER dependent signaling in mouse liver. Arch Biochem Biophys. 2007;459:273–279. doi: 10.1016/j.abb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 43.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2011;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zha BS, Zhou H. ER Stress and Lipid Metabolism in Adipocytes. Biochem Res Int. 2012;2012:312943. doi: 10.1155/2012/312943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10:1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J Immunol. 2006;176:6245–6253. doi: 10.4049/jimmunol.176.10.6245. [DOI] [PubMed] [Google Scholar]
- 47.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–243. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 49.Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, et al. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498–505. doi: 10.1152/ajpgi.00482.2007. [DOI] [PubMed] [Google Scholar]
- 50.Pfaffenbach KT, Gentile CL, Nivala AM, Wang D, Wei Y, Pagliassotti MJ. Linking endoplasmic reticulum stress to cell death in hepatocytes: roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am J Physiol Endocrinol Metab. 2010;298:E1027–1035. doi: 10.1152/ajpendo.00642.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soon RK, Jr, Yan JS, Grenert JP, Maher JJ. Stress signaling in the methionine-choline-deficient model of murine fatty liver disease. Gastroenterology. 2010;139:1730–1739. 1739, e1731. doi: 10.1053/j.gastro.2010.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, Friedman JE. CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–1117. doi: 10.1002/hep.21614. [DOI] [PubMed] [Google Scholar]
- 53.Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:159–166. doi: 10.1586/egh.11.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.