

Abstract
Methylamine dehydrogenase (MADH) requires the cofactor tryptophan tryptophylquinone (TTQ) for activity. TTQ is a posttranslational modification that results from an 8-electron oxidation of two specific tryptophans in the MADH β-subunit. The final 6-electron oxidation is catalyzed by an unusual c-type di-heme enzyme, MauG. The di-ferric enzyme can react with H2O2, but atypically for c-type hemes the di-ferrous enzyme can react with O2 as well. In both cases, an unprecedented bis-Fe(IV) redox state is formed, composed of a ferryl heme (Fe(IV)=O) and the second heme as Fe(IV) stabilized by His–Tyr axial ligation. Bis-Fe(IV) MauG acts as a potent 2-electron oxidant. Catalysis is long-range and requires a hole hopping electron transfer mechanism. This review highlights the current knowledge and focus of research into this fascinating system.
Introduction
Posttranslational modification of proteins evokes images of glycosylation, membrane anchors and signalling events, such as phosphorylation and methylation. However, it is becoming clear that the variety of posttranslational modifications extends far beyond these well-known examples. One such class is enzyme cofactors synthesized from amino acids within the active site.1–4 Many of the modifications involve residue crosslinking and/or oxidation to generate additional chemical reactivities beyond those of unmodified amino acids. Multiple examples of these enzymes also have transition metals in their active site, and many of the cofactors likely arose due to a side-reaction from activated species formed at the metal sites. Subsequently, some of these modified proteins evolved catalytic functions that required the chemically modified amino acids. This idea is borne out by the current crop of such modifications, which range from those that are not conserved with an unclear functional role (e.g. tyrosinase5–7), to those that have become a major player in the catalytic chemistry (e.g. KatG catalase activity8–13, copper amine oxidase14, 15) (Fig. 1). As would be expected, most of these are self-processed modifications that require the presence of the bound active site metal.8, 16–20 However, there are a few examples where the mature enzyme contains a protein-derived cofactor but no active site metal or other redox cofactor.21–23
Fig. 1.
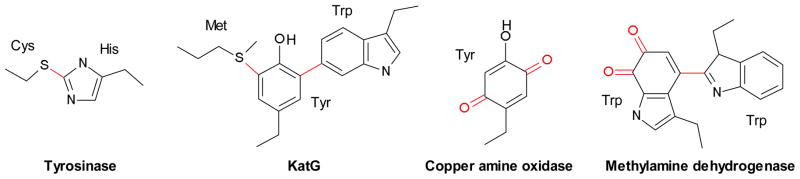
Posttranslationally modified amino acids found in four enzymes. Red indicates the modifications.
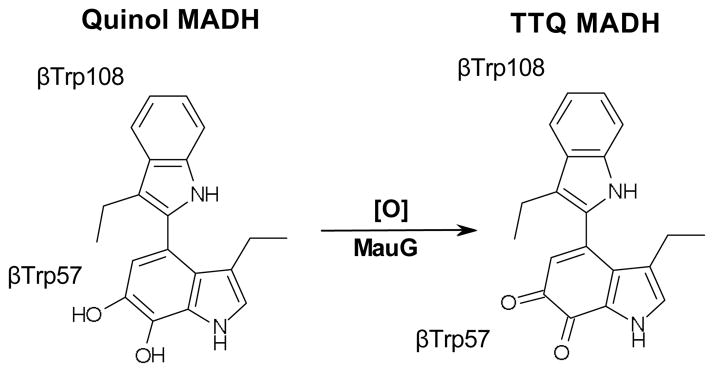
One such example is methylamine dehydrogenase (MADH) that contains the tryptophan tryptophylquinone cofactor (TTQ) (Fig. 1).22 MADH is a periplasmic enzyme found in a range of methylotrophic/autotrophic bacteria that can grow on methylamine as a sole source of energy and carbon. The enzyme catalyzes the 2-electron oxidation of methylamine to formaldehyde and ammonia.24 In Paracoccus denitrificans MADH is a 114 kDa α2β2 heterotetramer with two independent active sites, each containing a TTQ cofactor biosynthesized from βTrp57 and βTrp108 of the β-subunit.25, 26 TTQ biosynthesis is an 8-electron oxidation that adds two carbonyl oxygens to adjacent carbons in the indole ring of βTrp57, and covalently crosslinks βTrp57 to βTrp108. In P. denitrificans the methylamine utilization (mau) gene cluster contains 11genes, two of which encode the 42.5 kDa α-(mauB) and the 14.2 kDa β-subunits (mauA) of MADH.27 In knock-out studies, four genes were shown to be required for MADH maturation. The genes mauF28 and mauE29 are membrane proteins with no homology to characterized proteins, and are thought to be involved in transport of MADH subunits into the periplasm. Knocking out either gene leads to no detectable β-subunit in the periplasm, and an unusual β-subunit leader sequence is consistent with it being trafficked by a specific transporter. The loss of mauF and mauE additionally leads to a drastic reduction in α-subunit. The third gene, mauD, is homologous to di-sulfide isomerases, and is likely specific to the MADH β-subunit, which has six di-sulfides.29 In the absence of mauD, periplasmic α-subunit levels are close to normal, but again there is no detectable β-subunit implying that the di-sulfides are key to β-subunit stability. When the final required gene, mauG, is knocked out, there are normal levels of MADH α- and β-subunit in the periplasm, but no methylamine dehydrogenase activity is present.27 This has focused attention on the mauG gene product as a likely participant in TTQ biosynthesis.30, 31
MauG
MauG is undetectable in the P. denitrificans periplasm upon growth on methylamine, but can be homologously over-expressed.32 MauG contains two c-type heme binding sequence motifs (Cys-X-X-Cys-His) and the purified recombinant protein indeed contains two hemes. The enzyme is monomeric, with a molecular weight of 42 kDa. The EPR spectrum of di-ferric MauG shows the presence of equal amounts of low-spin (LS) and high-spin (HS) heme.32 Reduced di-ferrous MauG is able to form a stable complex with CO and reoxidizes in the presence of oxygen.32 This is surprising, as very few reduced c-type hemes react with exogenous, gaseous ligands. The MauG oxidation–reduction midpoint potentials (Em) for the addition of one electron and then a second are −159/−244 mV.33 However, resonance Raman and UV-visible absorbance demonstrated that both hemes reduce and oxidize simultaneously. Therefore, the hemes of MauG have similar Em but exhibit negative redox cooperativity, such that the first electron equilibrates between the two hemes making the addition of a second electron more difficult. As electrons are rapidly and efficiently transferred between hemes, MauG has a single di-heme redox cofactor and not two distinct sites of oxidation–reduction.
MauG catalytic activity
Recombinant and active MADH can be heterologously overexpressed in Rhodobacter sphaeroides by co-expression of the MADH structural genes, mauAB, in the presence of mauDEFG, the four genes required for MADH maturation.34 However, if mauAB are co-expressed with only mauDEF, α2β2 MADH protein is obtained that has no catalytic activity.35 Mass spectrometry demonstrated that this protein contains all six β-subunit di-sulfides, but has a partially synthesized TTQ cofactor with only a single –OH group added to the indole of βTrp57 (βTrp57-OH). Isotope labelling studies demonstrated that the hydroxyl group is at the C7 position on the indole ring.36 This precursor form of MADH is known as preMADH and the partially formed cofactor as preTTQ. The initial βTrp57 hydroxylation is a 2-electron oxidation, and the mechanism of this posttranslational modification is currently unknown.
C-type hemes are primarily involved in electron transfer (ET) processes through the use of the Fe(II)/Fe(III) redox couple. Some, such as the bacterial di-heme peroxidases that are MauG homologs, can catalytically use H2O2 in oxidative chemistry.37 However, MauG is highly unusual in that the di-ferrous c-type heme redox state can also bind and activate O2.32 Mixing preMADH with either di-ferric MauG and H2O2 or di-ferrous MauG and O2, leads to fully active MADH.38, 39 Thus, MauG represents the final enzyme in MADH maturation, catalyzing a 6-electron oxidation to complete TTQ biosynthesis (Fig. 2).
Fig. 2.
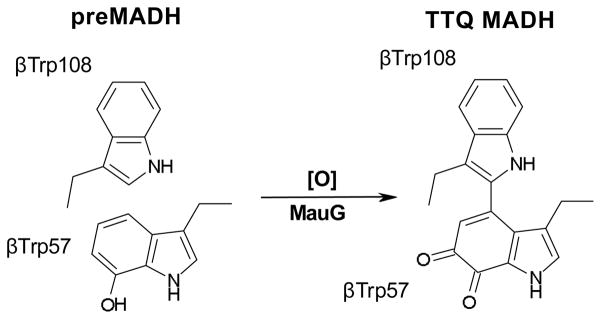
Paracoccus denitrificans MauG catalytic reaction.
Upon reaction of di-ferric MauG with H2O2, an unprecedented high-valent species is formed, which Mössbauer spectroscopy showed contains both MauG hemes in the Fe(IV) oxidation state.38 This bis-Fe(IV) redox form of MauG contains an Fe(IV)=O generated at the former HS heme (isomer shift = 0.06 mm/s, quadrupole splitting parameter = 1.70 mm/s), and an Fe(IV) heme coordinated by two amino acid ligands (isomer shift = 0.17 mm/s, quadrupole splitting parameter = 2.54 mm/s). Bis-Fe(IV) MauG decays slowly in air (k = 0.0002 s−1), but when mixed with preMADH it rapidly returns to the di-ferric state (k = 0.8 s−1).40 Furthermore, EPR shows the formation of a radical species concomitant with loss of bis-Fe(IV).38 The radical species is surprisingly long-lived, such that the reaction mixture can be resolved chromatographically, and the radical associated with the MADH protein. Further addition of H2O2 equivalents leads to TTQ formation, demonstrating that the bis-Fe(IV) MauG is catalytically competent. As bis-Fe(IV) MauG is reduced back to the di-ferric state by preMADH, this represents a 2-electron redox event. As MauG-dependent formation of active MADH is a 6-electron oxidation, there are three cycles of 2-electron oxidation events to complete TTQ biosynthesis requiring a total of three moles of either H2O2 or O2 plus reducing equivalents. These must equate to (1) insertion of a second –OH into βTrp57-OH, (2) crosslinking of βTrp57 and βTrp108, and (3) oxidation of the quinol to the quinone.
Although the order of the three 2-electron modifications is unknown, on the way to steady state an absorbance at 330 nm is transiently observed.41 This matches the λmax of the quinol form of MADH.42 Quinol can be generated by addition of dithionite to mature MADH, and further addition of stoichiometric bis-Fe(IV) MauG causes rapid oxidation to the quinone-containing resting state MADH.41 This provides evidence that oxidation of the quinol to the quinone is likely the last step in MauG-dependent TTQ formation.
Crystal structure of MauG in complex with preMADH
The crystal structure of MauG in complex with its protein substrate has been solved to 2.1 Å resolution (Fig. 3).43 As preMADH has two sites of TTQ formation, two MauG are symmetrically bound to each αβ heterodimer of the tetramer. PreMADH has essentially the same structure as mature MADH, with preTTQ βTrp108 coincident with its position in TTQ, and the plane of the βTrp57-OH indole rotated about 20° from its position in TTQ (Fig. 4). MauG consists of two structurally related domains, each of which contains a c-type heme. The heme closest to the interface is low-spin 6-coordinate with two amino acid ligands; the expected histidine axial ligand (His 205) from the CXXCH sequence motif of c-type hemes, and Tyr294 (Fig. 5A). The heme furthest from the interface is high-spin 5-coordinate with the expected histidine axial ligand (His 35) (Fig. 5B). The heme is accessible via a solvent channel, as would be expected for the site of H2O2/O2 binding. In addition, a recent molecular dynamics study using CO as a gas surrogate, suggests that there are multiple other paths for gas entry and exit at the distal side of the HS heme.44, 45 However, the distance between the preTTQ site and the iron of the HS heme is > 40 Å (Fig. 6), suggesting that either the structure is of an inactive form, or that the oxo of the bis-Fe(IV) MauG ferryl heme is not the source of the second TTQ oxygen. The latter was confirmed when H2O2 was added to the crystals and preTTQ was observed to convert to TTQ. Therefore, the crystal structure represents the catalytic enzyme-substrate complex, and the MauG catalytic mechanism must be very different from that of oxygenases and peroxidases.
Fig. 3.
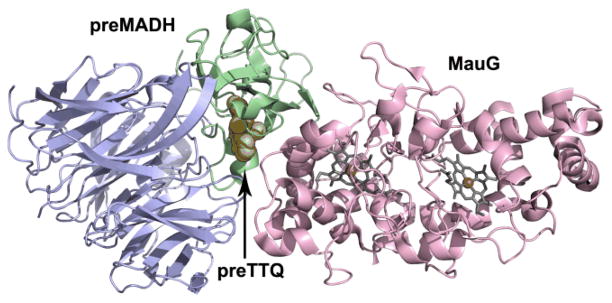
Overall fold of the complex between MauG (pink) and preMADH α-(blue) and β-subunits (green). Only half of preMADH is drawn. Heme porphyrins are drawn in grey stick with irons as orange spheres. PreTTQ is drawn in space-filling.
Fig. 4.

Key features of the preTTQ site (light green colored by atom). TTQ (carbon, dark green) from mature MADH is overlaid). MADH active site residues are drawn in stick colored by atom. The red sphere represents the only ordered water close to preTTQ. The dashed black line indicates a hydrogen bond.
Fig. 5.
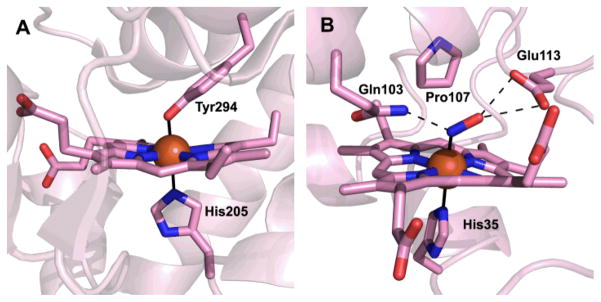
MauG hemes. (A) The 6-coordinate low-spin heme. (B) The 5-coordinate high-spin heme following reduction and complexation to nitric oxide. Heme porphyrins, amino acid ligands, residues in the HS heme distal pocket and NO are drawn in stick colored by atom. Irons are drawn as orange spheres. Solid black lines indicate axial iron ligation, and dashed lines indicate hydrogen bonds.
Fig. 6.

Proposed hole hopping pathway from preTTQ to the MauG hemes. Interaction distances in Angstroms are indicated by solid black lines. Molecular coloring as in Fig. 3. Heme porphyrins, Trp93, Trp199 and preTTQ are drawn in stick colored by atom. Irons are drawn as orange spheres. Calcium is drawn as a green sphere.
The MauG hemes have similar oxidation-reduction midpoint potentials, and so the redox cofactor is actually the entire di-heme unit. A conserved tryptophan (Trp93) is likely a key component of the highly efficient ET between the hemes, enabling rapid equilibration of electrons (Fig. 6). A small organic radical-like EPR signal (~ 1% of the protein) is associated with bis-Fe(IV) MauG that could represent oxidation of Trp93.38 Because the MauG redox cofactor is the entire di-heme unit, this reduces the distance between the potent MauG bis-Fe(IV) oxidant and preTTQ (19.4 Å from LS heme iron to closest preTTQ atom; 15.5 Å from LS heme porphyrin ring to preTTQ).43
When H2O2 was added to the MauG–preMADH crystals, electron density appeared at the open distal coordination site of the HS heme, further corroborating that this was the site of H2O2 binding and O2 activation.43 The crystal structure of the O2 structural mimic NO in complex with di-ferrous MauG–preMADH crystals confirmed that the distal side of the HS heme, which is furthest from the interface with preMADH, must be the site of O2/H2O2 binding and ferryl heme in bis-Fe(IV) MauG (Fig. 5B).45, 46 In the HS heme distal pocket, Glu113 is the only acid-base residue within a reasonable distance of the heme Fe, making it a likely candidate for the proton donor required for O-O bond cleavage. In support of this, the NO complex crystal structure shows that Glu113 directly interacts with the distal oxygen atom of the NO ligand, as well as a network of water molecules leading to bulk solvent. Gln103 also interacts with NO, donating a hydrogen bond to the proximal N atom. Based on its position and quantum chemical investigations,47 it is likely that Gln103 also interacts with the oxo ligand in the bis-Fe(IV) state, enhancing its stability. Finally, Pro107 has been shown to be important in maintaining the structure of the distal pocket in MauG, with mutations causing heme ligation by other residues and increased susceptibility to oxidation.48
MauG and bacterial di-heme cytochrome c peroxidases
It was already known from sequence alignments that MauG is related to bacterial di-heme cytochrome c peroxidases (bCCP).32, 37, 49 The closest bCCP sequence homolog to MauG that has been characterized is from Nitrosomonas europea (NeCCP), which shares 28% sequence identity with P. denitrificans MauG (Fig. 7).50, 51 However, the properties of the bCCP hemes differ significantly from those of MauG. In the bCCPs, the equivalent heme to the MauG LS heme (designated E-heme in bCCPs) has His–Met axial ligation rather than the His–Tyr ligands of MauG (Fig. 7B).37 In the oxidized form, most bCCPs are inactive and have His–His ligation at the equivalent MauG HS heme site (designated P-heme in bCCPs). The EPR spectrum of di-ferric MauG shows the presence of equal amounts of LS and HS heme, whereas most bCCPs are purely LS at 10K, due to the bis-His and His-Met coordination of their Fe(III) hemes. Reduced MauG is able to form a stable complex with CO that is observed to reoxidize in the presence of oxygen, but reduced bCCPs do not, the latter being typical behavior for c-type hemes.32 The oxidation–reduction midpoint potentials of MauG, which are very close and negative (−159/−244 mV),33 are also significantly different from those of the bCCPs, which are widely separated (−320/+320 mV for Pseudomonas aeruginosa).52 This reflects the fact that most bCCPs are only active in a mixed-valence state, where reduction of the His–Met ligated heme causes the His blocking the H2O2/O2 binding site to dissociate from the His–His ligated heme, converting it to HS and priming it for reaction with H2O2.53, 54 The Fe(III)/Fe(II) reacts with H2O2 to give ferryl heme, but the second oxidizing equivalent is stored as Fe(III) at the His–Met ligated heme. In MauG, however, a mixed-valence state is unobtainable, and the hemes act as a single redox unit.
Fig. 7.
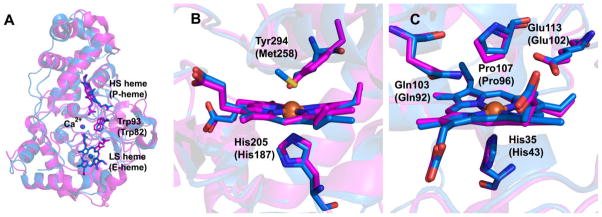
Overlays of the structures of MauG (magenta) and NeCCP (blue) comparing (A) the overall fold, (B) the LS heme and (C) the HS heme. Numbering for NeCCP is given in parentheses.
NeCCP is unique among the studied bCCPs in that both the di-ferric and mixed-valence states are active by virtue of the P-heme, which is HS and coordinated by a single His in both states (Fig. 7C).50, 51 The high-valent oxidant in the case of di-ferric NeCCP is Compound I, i.e. ferryl heme with the second oxidizing equivalent stored as a porphyrin-based radical cation. The distal residues of the HS heme are conserved between MauG and bCCPs, reflecting their common function in the binding and reduction of H2O2, and ability to stabilize Fe(IV)=O.
The major distinguishing feature of MauG that accounts for its unusual properties is found at the LS heme. This heme is ligated by His205 and Tyr294, and is the only example of a c-type heme with such a ligand set (Fig. 5A).43 This probably accounts for the dramatic decrease in reduction potential relative to bCCPs, as the incorporation of an anionic tyrosinate ligand would be expected to favor higher oxidation states. As such, it is also required to stabilize the Fe(IV) oxidation state of the heme in the MauG bis-Fe(IV) intermediate. In support of this, the mutation of Tyr294 to His in MauG converts the LS heme to bis-His coordination, altering the reduction potentials of both hemes and making it unable to form the bis-Fe(IV) intermediate upon reaction with H2O2.55 Rather, a Compound I-like species is formed, indicating that all oxidizing potential is centered on the HS heme and heme cooperativity is effectively severed.
A final difference is that bCCPs are active as homodimers, but MauG is monomeric.37 Dimerization and activation of bCCPs are often linked to calcium binding, with a conserved calcium site present in all bCCPs and MauG (Fig. 7A). This sits at the interface of the two heme-bearing domains near Trp93 of MauG, which lies equidistant between the hemes (this Trp is also strictly conserved in bCCPs). In addition, bCCPs have a second Ca2+ binding site formed at the dimer interface. In MauG, when Ca2+ is depleted circular dichroism shows that the secondary structure remains intact.56 Depletion of Ca2+ from MauG leads to altered heme properties and inactive enzyme that cannot form bis-Fe(IV). This effect is completely reversible upon addition of calcium. Therefore, the Ca2+ seems to be important for maintaining close association of the hemes through restriction of domain-domain dynamics. Resonance Raman and EPR spectroscopies show that the HS heme of MauG is converted to LS in Ca2+-depleted MauG,56 and EPR and Mössbauer spectroscopies show the axial ligand orientation at the His–Tyr LS heme changes.57 Ca2+-depleted MauG can still effect a non-physiological, but thermodynamically favorable, ET reaction from di-ferrous MauG to MADH, which does not require the bis-Fe(IV) redox state, to yield quinol MADH and di-ferric MauG, demonstrating that Ca2+-depleted MauG can still bind to MADH (Fig. 8C). 56
Fig. 8.

Single turnover reactions of MauG that have been kinetically characterized.
MauG kinetics
To complete TTQ synthesis from preTTQ, MauG must undergo three cycles of 2-electron oxidation events using the bis-Fe(IV) intermediate to oxidize preMADH. The order of modifications brought about by the 2-electron oxidations is unknown, these being crosslink formation, –OH insertion and oxidation of quinol to quinone. The catalytic competence of the MauG–preMADH crystals means that MauG-dependent synthesis of TTQ is processive (MauG does not need to dissociate between the 2-electron oxidations) and that no significant conformational changes occur during turnover.43
A number of different steady state and single turnover reactions have been studied kinetically. The overall reaction has steady state parameters of kcat = 0.2 s−1 and Km = 6.6 μM, irrespective of whether H2O2 or O2 plus reductant is used as a substrate (Fig. 2).58 The initial 2-electron oxidation has also been studied in single turnover experiments (Fig. 8A).40 Making use of the unusual longevity of the bis-Fe(IV) state of MauG (rate of formation > 300 s−1; rate of spontaneous decay = 0.0002 s−1) and the ability of di-ferric MauG to form a complex with preMADH (Kd ≤ 1.5 μM), it was shown that the order of addition of substrate preMADH or stoichiometric addition of H2O2/reductant + O2 made no difference to the kinetic parameters of the initial 2-electron MauG-dependent oxidation (limiting first-order rate constant (klim) = 0.80 s−1), indicating a random order mechanism. This is unusual as other reactions that make use of high-valent hemes, such as Compound I of cytochromes P450, have ordered mechanisms in which the heme cannot activate O2 until the organic substrate is bound.59–61 This has likely evolved due to the normally rapid decay of high-valent species that often leads to free radical-initiated oxidative damage. Although the spontaneous decay of MauG bis-Fe(IV) is slow, the enzyme does inactivate upon repeated rounds of bis-Fe(IV) formation and decay (i.e. in the absence of preMADH), and MauG is essentially inactive after 4 cycles of treatment.62 The presence of hydroxyurea, a radical scavenger, protects MauG from inactivation, with little loss of activity after 12 cycles of bis-Fe(IV) formation and decay, consistent with a free radical mechanism leading to oxidative damage.
As mentioned earlier, the final 2-electron oxidation is thought to be the quinol to quinone oxidation, as a transient intermediate is observed during attainment of steady state with an absorbance equivalent to dithionite reduced mature MADH (λmax = 330 nm).41, 42 Mature MADH reduced by dithionite is a substrate of MauG in the presence of H2O2, with steady state parameters of kcat = 4.1 s−1 and Km = 11.1 μM (Fig. 9).63 This reaction can also be studied under single turnover conditions through stoichiometric reduction of MADH by dithionite followed by mixing with preformed bis-Fe(IV) MauG (Fig. 8B). In this case the reaction exhibits a klim = 20 s−1 and Kd = 11.2 μM.63 The kcat for this reaction and the klim under single turnover conditions are both over 20-fold higher than for the overall reaction and the first 2-electron oxidation event, suggesting that this oxidation is not rate-limiting in MauG-dependent TTQ biosynthesis.40, 58 MauG-dependent quinol to quinone oxidation in MADH also requires formation of the MauG bis-Fe(IV) redox state, as shown by studies involving mutation of the unusual LS heme ligand Tyr294 to His.55 Upon addition of H2O2, this MauG variant forms an Fe(V)-equivalent intermediate, but resonance Raman and UV-visible absorption spectroscopies show that the second oxidizing equivalent is restricted to the area of the ferryl heme, and Y294H MauG cannot catalyze TTQ formation from either preMADH or quinol MADH.
Fig. 9.
Steady state reaction of MauG-dependent oxidation of quinol to quinone MADH thought to be the final step in TTQ maturation.
Figure 10 summarizes the current knowledge regarding MauG-dependent TTQ maturation.
Fig. 10.

Current reaction scheme for MauG-dependent TTQ biosynthesis.
MauG-dependent completion of TTQ biosynthesis requires a hole hopping electron transfer mechanism
The oxidation of di-ferrous MauG by mature quinone MADH is thermodynamically favorable, and mixing of these two species leads to rapid formation of di-ferric MauG and quinol MADH (klim = 0.7 s−1, Kd = 10.1 μM) (Fig. 8C).63 This non-physiological, single turnover ET reaction, which does not involve the MauG bis-Fe(IV) redox state, has been used to check whether MauG loss-of-function perturbations, such as variants resulting from site-directed mutagenesis55, 64 or Ca2+ depletion56, are a consequence of disrupted binding between MauG and preMADH/quinol MADH.
In the case of the MauG Y294H variant, the inability to catalyze TTQ biosynthesis was not the result of the proteins being unable to interact, as MADH could oxidize di-ferrous Y294H MauG (klim = 0.21 s−1, Kd = 9.4 μM) (Fig. 8C).55 As the Y294H MauG high-valent species is restricted to the vicinity of the heme furthest from preMADH, successful MauG catalysis must depend on the formation of the bis-Fe(IV) redox state. Although the site of the Fe(IV)=O is 40.1 Å from the closest atom of preTTQ, the redox cooperativity between the hemes means that the distance between the bis-Fe(IV) oxidant and the closest preTTQ atom is effectively reduced to 15.5 Å (to the LS heme porphyrin; 14.0 Å to the closest LS heme atom) (Fig. 6). Thus, MauG provides a long-range driving force, and the chemistry at the preTTQ site is being directed by the MADH active site residues, which are identically arranged to that seen in mature MADH (Fig. 4). Thus, these residues likely play a role in two very different chemistries: TTQ biosynthesis and methylamine oxidation. This multi-functionality is also a common theme in self-processed protein-derived cofactor biosynthesis.65 As the catalysis is long-range, the source of the second TTQ oxygen is probably solvent, unlike other Fe(V)-equivalent heme-based oxidant systems, such as cytochromes P450 and peroxidases, where it is the oxo of ferryl heme.61, 66 In MauG, the ferryl oxo is more than 40 Å from the site of oxygen addition to preMADH βTrp57-OH.
However, the distance is still significant between the LS heme and preTTQ, and the kcat for TTQ biosynthesis (0.2 s−1) is too fast for a mechanism involving ET tunneling across such a distance. However, there is a MauG Trp residue (Trp199) equidistant between the LS heme of MauG and preTTQ of preMADH (Fig. 6). This residue was mutated to Phe and Lys, neither of which can be oxidized.64 Both W199F and W199K MauG can form the bis-Fe(IV) intermediate, but neither can carry out TTQ biosynthesis with preMADH as substrate. This strongly suggested that the distance between the LS heme and preTTQ was being bridged by hole hopping through Trp199, effectively shortening the ET distance. As Trp199 lies at the interface between MauG and preMADH, it was particularly important to demonstrate that the W199F/K MauG variants could still bind to preMADH with minimal perturbation of the interprotein interaction. This was demonstrated to be the case both by the oxidation of di-ferrous W199F/K MauG by MADH, and the determination of crystal structures of the variants in complex with preMADH that were essentially identical to the wild-type MauG–preMADH complex, except at the site of mutation. Furthermore, although addition of H2O2 to crystals of wild-type MauG–preMADH crystals leads to TTQ biosynthesis in crystallo, this does not occur in the case of the Trp199 variant-containing crystals. Interestingly, the MauG-dependent 2-electron oxidation of quinol to quinone MADH was supported, although with a kcat/Km value an order of magnitude lower than wild-type MauG (W199F MauG: kcat = 0.55 s−1, Km = 25.6 μM) (Fig. 9). It appears that in this case, the driving force created by the bis-Fe(IV) is strong enough to enable this oxidation to proceed by long-range electron tunneling, whereas it cannot for the first 2-electron oxidation of MauG-dependent TTQ biosynthesis (Fig. 8A). These results have recently been corroborated by temperature dependence kinetic studies on the single-turnover rates of ET from preMADH to wild-type MauG bis-Fe(IV) and from di-ferrous wild-type MauG to preMADH, coupled to Marcus analysis.67, 68 In conclusion, both Trp93 and Trp199 of MauG decrease the ET distance between the ferryl heme and preTTQ, the former through enabling redox cooperativity between the MauG hemes to effectively give a di-heme redox cofactor, and the latter by supporting a multistep hole hopping mechanism between the LS heme of MauG and preTTQ.
Conclusions
The MauG posttranslational machinery gives remarkable insight into how a protein matrix controls the stabilization of Fe(V)-equivalent heme-based oxidants and long-range interprotein electron transfer. In particular, there are few biological systems where good evidence for a multistep ET hopping mechanism has been obtained. These include DNA,69 Zn-substituted cytochrome c peroxidase,70 ribonucleotide reductase,71 DNA photolyase72 and a Rh-labeled azurin73. The stabilization of the Fe(V)-equivalent MauG bis-Fe(IV) is unprecedented, and enables the study of free radical pathways and oxidative damage mechanisms within a protein matrix. The ability to probe distinct ET reactions within the same protein system is leading to general rules regarding electron movement in biology. There are exciting times ahead with regard MauG research, as we increase our understanding of ET within proteins (the basis of metabolism), oxidative damage and stabilization of high-valent iron species.
Acknowledgments
This work was supported by NIH grants GM-66569 (CMW) and GM-97779 (ETY). Figures were generated using resources in the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute.
Biographies
 Carrie Wilmot received her PhD from Birkbeck College in 1989 working with Janet Thornton. She was a Cancer Research Institute Postdoctoral Fellow with Ian Wilson at The Scripps Research Institute, La Jolla, USA. Returning to the University of Leeds, her undergraduate alma mater, to work with Simon Phillips, she began her research into understanding the catalytic chemistry and biosynthesis of protein-derived cofactors. As an EMBO Short-Term Fellow working with Janos Hajdu at Uppsala University, Sweden, she began to combine crystallography with single crystal spectroscopy. She is now Professor of Biochemistry at the University of Minnesota, USA.
Carrie Wilmot received her PhD from Birkbeck College in 1989 working with Janet Thornton. She was a Cancer Research Institute Postdoctoral Fellow with Ian Wilson at The Scripps Research Institute, La Jolla, USA. Returning to the University of Leeds, her undergraduate alma mater, to work with Simon Phillips, she began her research into understanding the catalytic chemistry and biosynthesis of protein-derived cofactors. As an EMBO Short-Term Fellow working with Janos Hajdu at Uppsala University, Sweden, she began to combine crystallography with single crystal spectroscopy. She is now Professor of Biochemistry at the University of Minnesota, USA.
 Erik Yukl received a BS in Chemistry from Pacific University, Oregon, USA, in 2005. He completed his PhD in 2009 at Oregon Health and Science University, USA, working with Pierre Moënne-Loccoz on NO-sensing and detoxification by bacterial iron-containing proteins, and published 13 manuscripts in the process. Continuing his love of iron, he is currently a National Institutes of Health NRSA Postdoctoral Fellow at the University of Minnesota, USA, interrogating the MauG system with crystallography, single crystal spectroscopies and mass spectrometry.
Erik Yukl received a BS in Chemistry from Pacific University, Oregon, USA, in 2005. He completed his PhD in 2009 at Oregon Health and Science University, USA, working with Pierre Moënne-Loccoz on NO-sensing and detoxification by bacterial iron-containing proteins, and published 13 manuscripts in the process. Continuing his love of iron, he is currently a National Institutes of Health NRSA Postdoctoral Fellow at the University of Minnesota, USA, interrogating the MauG system with crystallography, single crystal spectroscopies and mass spectrometry.
Notes and references
- 1.Davidson VL. Biochemistry. 2007;46:5283–5292. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 2.Davidson VL. Mol BioSyst. 2011;7:29–37. doi: 10.1039/c005311b. [DOI] [PubMed] [Google Scholar]
- 3.Okeley NM, van der Donk WA. Chem & Biol. 2000;7:R159–171. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 4.Yukl ET, Wilmot CM. Curr Opin Chem Biol. 2012;16:54–59. doi: 10.1016/j.cbpa.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claus H, Decker H. Syst Appl Microbiol. 2006;29:3–14. doi: 10.1016/j.syapm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Borron JC, Solano F. Pigment Cell Res. 2002;15:162–173. doi: 10.1034/j.1600-0749.2002.02012.x. [DOI] [PubMed] [Google Scholar]
- 7.Lerch K. Mol Cell Biochem. 1983;52:125–138. doi: 10.1007/BF00224921. [DOI] [PubMed] [Google Scholar]
- 8.Ghiladi RA, Knudsen GM, Medzihradszky KF, de Montellano PRO. J Biol Chem. 2005;280:22651–22663. doi: 10.1074/jbc.M502486200. [DOI] [PubMed] [Google Scholar]
- 9.Ghiladi RA, Medzihradszky KF, de Montellano PRO. Biochemistry. 2005;44:15093–15105. doi: 10.1021/bi051463q. [DOI] [PubMed] [Google Scholar]
- 10.Jakopitsch C, Auer M, Ivancich A, Ruker F, Furtmuller PG, Obinger C. J Biol Chem. 2003;278:20185–20191. doi: 10.1074/jbc.M211625200. [DOI] [PubMed] [Google Scholar]
- 11.Jakopitsch C, Ivancich A, Schmuckenschlager F, Wanasinghe A, Poltl G, Furtmuller PG, Ruker F, Obinger C. J Biol Chem. 2004;279:46082–46095. doi: 10.1074/jbc.M408399200. [DOI] [PubMed] [Google Scholar]
- 12.Yu SW, Girotto S, Zhao XB, Magliozzo RS. J Biol Chem. 2003;278:44121–44127. doi: 10.1074/jbc.M304757200. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, Khajo A, Jarrett S, Suarez J, Levitsky Y, Burger RM, Jarzecki AA, Magliozzo RS. J Biol Chem. doi: 10.1074/jbc.M112.401208. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanizawa K, Matsuzaki R, Shimizu E, Yorifuji T, Fukui T. Biochem Biophys Res Comm. 1994;199:1096–1102. doi: 10.1006/bbrc.1994.1343. [DOI] [PubMed] [Google Scholar]
- 15.Wilmot CM, Murray JM, Alton G, Parsons MR, Convery MA, Blakeley V, Corner AS, Palcic MM, Knowles PF, McPherson MJ, Phillips SE. Biochemistry. 1997;36:1608–1620. doi: 10.1021/bi962205j. [DOI] [PubMed] [Google Scholar]
- 16.Bollinger JA, Brown DE, Dooley DM. Biochemistry. 2005;44:11708–11714. doi: 10.1021/bi0504310. [DOI] [PubMed] [Google Scholar]
- 17.Cai D, Klinman JP. J Biol Chem. 1994;269:32039–32042. [PubMed] [Google Scholar]
- 18.Fujieda N, Ikeda T, Murata M, Yanagisawa S, Aono S, Ohkubo K, Nagao S, Ogura T, Hirota S, Fukuzumi S, Nakamura Y, Hata Y, Itoh S. J Amer Chem Soc. 2011;133:1180–1183. doi: 10.1021/ja108280w. [DOI] [PubMed] [Google Scholar]
- 19.Matsuzaki R, Fukui T, Sato H, Ozaki Y, Tanizawa K. FEBS Lett. 1994;351:360–364. doi: 10.1016/0014-5793(94)00884-1. [DOI] [PubMed] [Google Scholar]
- 20.Whittaker MM, Whittaker JW. J Biol Chem. 2003;278:22090–22101. doi: 10.1074/jbc.M300112200. [DOI] [PubMed] [Google Scholar]
- 21.Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, Mathews FS. Proc Natl Acad Sci USA. 2001;98:14268–14273. doi: 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. Science. 1991;252:817–824. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 23.Satoh A, Kim JK, Miyahara I, Devreese B, Vandenberghe I, Hacisalihoglu A, Okajima T, Kuroda S, Adachi O, Duine JA, Van Beeumen J, Tanizawa K, Hirotsu K. J Biol Chem. 2002;277:2830–2834. doi: 10.1074/jbc.M109090200. [DOI] [PubMed] [Google Scholar]
- 24.Davidson VL. Bioorg Chem. 2005;33:159–170. doi: 10.1016/j.bioorg.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Chen LY, Doi N, Durley RCE, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 26.Chen LY, Mathews FS, Davidson VL, Huizinga EG, Vellieux FM, Duine JA, Hol WG. FEBS Lett. 1991;287:163–166. doi: 10.1016/0014-5793(91)80041-z. [DOI] [PubMed] [Google Scholar]
- 27.van der Palen CJ, Slotboom DJ, Jongejan L, Reijnders WN, Harms N, Duine JA, van Spanning RJ. Eur J Biochem. 1995;230:860–871. doi: 10.1111/j.1432-1033.1995.tb20629.x. [DOI] [PubMed] [Google Scholar]
- 28.Chistoserdov AY, Boyd J, Mathews FS, Lidstrom ME. Biochem Biophys Res Comm. 1992;184:1181–1189. doi: 10.1016/s0006-291x(05)80007-5. [DOI] [PubMed] [Google Scholar]
- 29.van der Palen CJ, Reijnders WN, deVries S, Duine JA, van Spanning RJ. Antonie van Leewenhoek. 1997;72:219–228. doi: 10.1023/a:1000441925796. [DOI] [PubMed] [Google Scholar]
- 30.Davidson VL, Liu A. Biochim Biophys Acta. 2012;1824:1299–1305. doi: 10.1016/j.bbapap.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilmot CM, Davidson VL. Curr Opin Chem Biol. 2009;13:469–474. doi: 10.1016/j.cbpa.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Graichen ME, Liu A, Pearson AR, Wilmot CM, Davidson VL. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Biochemistry. 2006;45:821–828. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graichen ME, Jones LH, Sharma BV, van Spanning RJM, Hosler JP, Davidson VL. J Bacteriol. 1999;181:4216–4222. doi: 10.1128/jb.181.14.4216-4222.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pearson AR, De La Mora-Rey T, Graichen ME, Wang Y, Jones LH, Marimanikkupam S, Agger SA, Grimsrud PA, Davidson VL, Wilmot CM. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 36.Pearson AR, Marimanikkuppam S, Li X, Davidson VL, Wilmot CM. J Amer Chem Soc. 2006;128:12416–12417. doi: 10.1021/ja064466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettigrew GW, Echalier A, Pauleta SR. J Inorg Biochem. 2006;100:551–567. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Fu R, Lee S, Krebs C, Davidson VL, Liu A. Proc Natl Acad Sci USA. 2008;105:8597–8600. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. J Amer Chem Soc. 2005;127:8258–8259. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 40.Lee S, Shin S, Li X, Davidson VL. Biochemistry. 2009;48:2442–2447. doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Biochemistry. 2006;45:13276–13283. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 42.Davidson VL, Brooks HB, Graichen ME, Jones LH, Hyun YL. Meth Enzymol. 1995;258:176–190. doi: 10.1016/0076-6879(95)58046-8. [DOI] [PubMed] [Google Scholar]
- 43.Jensen LM, Sanishvili R, Davidson VL, Wilmot CM. Science. 2010;327:1392–1394. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pietra F. Chem Biodiversity. 2012;9:1425–1435. doi: 10.1002/cbdv.201200176. [DOI] [PubMed] [Google Scholar]
- 45.Yukl ET, Goblirsch BR, Davidson VL, Wilmot CM. Biochemistry. 2011;50:2931–2938. doi: 10.1021/bi200023n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu R, Liu F, Davidson VL, Liu A. Biochemistry. 2009;48:11603–11605. doi: 10.1021/bi9017544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ling Y, Davidson VL, Zhang Y. J Phys Chem Lett. 2010;1:2936–2939. doi: 10.1021/jz101159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feng ML, Jensen LMR, Yukl ET, Wei XX, Liu AM, Wilmot CM, Davidson VL. Biochemistry. 2012;51:1598–1606. doi: 10.1021/bi201882e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atack JM, Kelly DJ. Adv Microb Physiol. 2007;52:52, 73–106. doi: 10.1016/S0065-2911(06)52002-8. [DOI] [PubMed] [Google Scholar]
- 50.Arciero DM, Hooper AB. J Biol Chem. 1994;269:11878–11886. [PubMed] [Google Scholar]
- 51.Shimizu H, Schuller DJ, Lanzilotta WN, Sundaramoorthy M, Arciero DM, Hooper AB, Poulos TL. Biochemistry. 2001;40:13483–13490. doi: 10.1021/bi011481h. [DOI] [PubMed] [Google Scholar]
- 52.Ellfolk N, Ronnberg M, Aasa R, Andreasson LE, Vanngard T. Biochim Biophys Acta. 1983;743:23–30. doi: 10.1016/0167-4838(83)90413-2. [DOI] [PubMed] [Google Scholar]
- 53.Echalier A, Brittain T, Wright J, Boycheva S, Mortuza GB, Fulop V, Watmough NJ. Biochemistry. 2008;47:1947–1956. doi: 10.1021/bi702064f. [DOI] [PubMed] [Google Scholar]
- 54.Echalier A, Goodhew CF, Pettigrew GW, Fulop V. Structure. 2006;14:107–117. doi: 10.1016/j.str.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 55.Abu Tarboush N, Jensen LMR, Feng ML, Tachikawa H, Wilmot CM, Davidson VL. Biochemistry. 2010;49:9783–9791. doi: 10.1021/bi101254p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin S, Feng M, Chen Y, Jensen LM, Tachikawa H, Wilmot CM, Liu A, Davidson VL. Biochemistry. 2011;50:144–150. doi: 10.1021/bi101819m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Naik SG, Krzystek J, Shin S, Nelson WH, Xue S, Yang JJ, Davidson VL, Liu A. Biochemistry. 2012;51:1586–1597. doi: 10.1021/bi201575f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X, Fu R, Liu A, Davidson VL. Biochemistry. 2008;47:2908–2912. doi: 10.1021/bi702259w. [DOI] [PubMed] [Google Scholar]
- 59.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 60.Makris TM, von Koenig K, Schlichting I, Sligar SG. J Inorg Biochem. 2006;100:507–518. doi: 10.1016/j.jinorgbio.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 61.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 62.Shin S, Lee S, Davidson VL. Biochemistry. 2009;48:10106–10112. doi: 10.1021/bi901284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shin S, Abu Tarboush N, Davidson VL. Biochemistry. 2010;49:5810–5816. doi: 10.1021/bi1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abu Tarboush N, Jensen LMR, Yukl ET, Geng JF, Liu AM, Wilmot CM, Davidson VL. Proc Natl Acad Sci USA. 2011;108:16956–16961. doi: 10.1073/pnas.1109423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klinman JP. Biochim Biophys Acta. 2003;1647:131–137. doi: 10.1016/s1570-9639(03)00077-3. [DOI] [PubMed] [Google Scholar]
- 66.Gumiero A, Metcalfe CL, Pearson AR, Raven EL, Moody PC. J Biol Chem. 2010;286:1260–1268. doi: 10.1074/jbc.M110.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 68.Choi M, Shin S, Davidson VL. Biochemistry. 2012;51:6942–6949. doi: 10.1021/bi300817d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Genereux JC, Barton JK. Chem Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seifert JL, Pfister TD, Nocek JM, Lu Y, Hoffman BM. J Amer Chem Soc. 2005;127:5750–5751. doi: 10.1021/ja042459p. [DOI] [PubMed] [Google Scholar]
- 71.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 72.Lukacs A, Eker APM, Byrdin M, Brettel K, Vos MH. J Amer Chem Soc. 2008;130:14394–14395. doi: 10.1021/ja805261m. [DOI] [PubMed] [Google Scholar]
- 73.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlcek A, Richards JH, Winkler JR, Gray HB. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]