

Abstract
We previously reported that peripheral leptin infusions in chronically decrebrate rats, in which the forebrain is neurally isolated from the hindbrain, increased body fat and decreased energy expenditure. Any central leptin response in decerebrate rats would depend upon the hindbrain. Here, we tested whether selective activation of hindbrain leptin receptors increased body fat. Fourth ventricle infusion of 0.6 μg leptin/day for 12 days increased body fat by 13% with no increase in food intake. Third ventricle leptin infusions decreased food intake, body fat, and lean tissue with a maximal response at 0.3 μg leptin/day. To test whether hindbrain receptors opposed activity of hypothalamic receptors, rats received peripheral infusions of 40 μg leptin/day and increasing 4th ventricle doses of the leptin receptor antagonist mutein protein. Mutein (3.0 μg/day) reduced body fat in PBS-infused rats to the same level as leptin-infused rats and reduced lean tissue in all rats. Leptin, but not mutein, inhibited food intake. By contrast, 3.0 μg/day mutein in the 3rd ventricle increased food intake and body fat in both PBS- and leptin-infused rats. In basal conditions, hindbrain leptin receptors may antagonize activity of forebrain receptors to protect lean and fat tissue, but there is no evidence for an anabolic role for hindbrain receptors when leptin is elevated. In a dietary study, rats increased energy intake when offered lard and 30% sucrose solution in addition to chow. Peripheral leptin infusion exaggerated the gain in body fat without altering energy intake confirming the potential for leptin to increase adiposity.
Keywords: forebrain, hindbrain, mutein leptin, body composition
leptin is an adipose-derived hormone that is hypothesized to be a negative feedback signal in the regulation of energy balance (46). Central (12, 43) or peripheral (37) administration of leptin inhibits food intake of normal-weight rodents and corrects many aspects of the obesity syndrome in leptin-deficient ob/ob mice (20, 33). The negative energy balance associated with peripheral leptin treatment results in a specific loss of body fat mass and maintenance of lean body mass (20); by contrast, central administration of leptin may result in the loss of both fat and lean tissue (5). Even though leptin is an effective regulator of energy balance in normal-weight rodents, obese or aged animals have high circulating concentrations of endogenous leptin but are leptin resistant because leptin administration does not influence food intake or body weight (7, 9, 44, 45).
Early observations that peripheral leptin resistance preceded the development of central leptin resistance in high-fat-fed mice (9) led to the conclusion that peripheral leptin resistance results from a failure of leptin to cross the blood-brain barrier (1), whereas central leptin resistance is a failure of leptin to activate its receptors (38). Consistent with these observations, leptin resistance in diet-induced obese rats is associated with a failure to activate hypothalamic receptors, while hindbrain receptors remain leptin responsive (25). Others (29), however, have reported that leptin receptors in only specific areas of the hypothalamus become unresponsive in leptin-resistant diet-induced obese mice, suggesting that the resistance is associated with changes in receptor sensitivity rather than transport of leptin across the blood-brain barrier.
Because circulating leptin can cross the blood-brain barrier, many of the responses to peripheral leptin result from activation of centrally located receptors (4, 35). Leptin injected into the third ventricle, however, causes an elevation of circulating leptin concentrations (26, 32, 34). Thus, when leptin is administered to an experimental animal, it is often difficult to discern the site of leptin activity. In a previous effort to clearly define the importance of forebrain neural efferent signals in mediating energetic responses to peripherally administered leptin, we tested the effects of a peripheral infusion of a physiological concentration of leptin in chronic decerebrate (CD) rats (18). Surgical transection of the brain in CD rats destroys the neural communication between the forebrain and the rest of the body, including the hindbrain (22); therefore, any response to peripheral leptin has to be mediated by humoral communication between the forebrain and the rest of the body, neural output from the hindbrain, and/or direct effects of leptin in the periphery. We were surprised to find that peripherally infused leptin caused a small, but significant, increase in body fat content of CD rats even though they were already significantly fatter than their intact controls (18). Because the CD rats were tube fed, the increase in body fat mass was independent of a change in food intake, but energy expenditure was inhibited during the light phase in leptin-treated CD rats (18). The increase in body fat of leptin-treated rats is not limited to CD rats, as we had previously reported that a 12-day peripheral infusion of a physiological dose of leptin in mice fed a high-fat diet and housed in a warm environment increased body fat (19). We proposed that, although the high-fat diet prevented a leptin-induced inhibition of food intake, these mice were not totally leptin resistant and, in specific situations when leptin activity in the forebrain was inhibited, body fat would increase due to an inhibition of energy expenditure.
The data from the CD rats led us to further hypothesize that in nonstimulated conditions leptin receptors located in the hindbrain and/or periphery antagonize the catabolic effect of leptin activity in the forebrain. The objective of the studies described here was to test whether forebrain and hindbrain leptin receptors have different effects on body composition. This was accomplished by chronically infusing low doses of leptin into either the 3rd or the 4th ventricle of rats or by infusing the rats peripherally with leptin while inhibiting activation of forebrain or hindbrain receptors by infusion of the leptin receptor antagonist leptin mutein protein (mutein) (42) into the 3rd or 4th ventricle.
METHODS
Rats used in these studies were male Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN) housed with lights on for 12 h each day from 0700. They were individually housed in wire mesh cages. All animals had free access to chow (LabDiet 5012; PMI Nutrition International, Brentwood, MO) and water unless stated otherwise. Each rat had a Nylabone (Nylabone Products, Neptune, NJ) in their cage for enrichment. All animal procedures were approved by the Institutional Animal Care Use Committees of University of Georgia and of Georgia Health Sciences University.
Experiment 1: effect of a 4th ventricle low-dose leptin infusion.
Because chronically decerebrate rats increased their body fat mass in response to a peripheral infusion of leptin (18), this study tested whether specific activation of leptin receptors in the hindbrain by low doses of leptin would cause a similar change in body composition. Thirty-five rats, housed as described above in a room maintained at 23°C, were each fitted with a 4th ventricle infusion cannula (Plastics One, Roanoake, VA) as described previously (27). The coordinates in relation to the midline at the occipital suture were 2.5 mm anterior, 0 mm lateral, and −6.5 mm ventral. They were allowed 1 wk to recover from the surgery and weighed 400–420 g at the start of the experiment. Baseline daily body weights and food intakes were recorded for 5 days before the rats were divided into three weight-matched groups. With the rats under isoflurane anesthesia, an Alzet miniosmotic pump (model 1002; Durect, Cupertino, CA) was placed subcutaneously and connected to the cannula to deliver PBS, 0.15 μg leptin/24 h or 0.6 μg leptin/24 h into the 4th ventricle for 12 days in an infusion volume of 0.25 μl/h. PBS was used for the control infusion because this was the vehicle for the leptin. At the same time that the pump was placed, the rats were fitted with a Thermicron iButton (DS1922L; Embedded Data Systems, Lawrenceburg, KY) that was set to record temperature every 30 min with a precision of 0.06°C starting 24 h after pump placement and continuing until the end of the study. The iButton was placed underneath the intrascapular brown fat (IBAT) for one-half of the rats in each group and intraperitoneally (ip) for the other half.
On day 12 of infusion, food was removed from the rat cages at 7:00 AM. Starting at 10:00 AM, rats were killed, and trunk blood was collected for measurement of serum leptin (Rat Leptin RIA kit, Millipore). Inguinal, epididymal, retroperitoneal, and mesenteric white fat and IBAT were dissected, weighed, and returned to the carcass. Carcass fat was determined as described previously (15).
An additional 14 rats were fitted with Alzet pumps to deliver either PBS or 0.6 μg leptin/24 h into the 4th ventricle. At the end of 5 days of infusion, two rats per group were food deprived from 7:00 AM to 12:00 noon before being perfused with heparinized saline followed by 4% paraformaldehyde. The brains were collected and left in 4% paraformaldehyde overnight and then stored in sucrose azide solution. Coronal 30-μm sections were made, and hypothalamic and brain stem phosphorylated signal transducer and activator of transcription-3 (pSTAT3) was detected by immunohistochemistry as described previously (14). The remaining 10 rats (5 per group) were food deprived from 7:00 AM to 11:00 AM, and tissue blocks of the hypothalamus or brain stem were collected for measurement of phosphorylated STAT3, phosphoinositide 3-kinase (pPI3K p85), extracellular signal-related kinases 1/2 (pERK1, pERK2), and AMP-activated protein kinase (pAMPK), suppressor of cytokine signaling 3 (SOCS3), and protein-tyrosine phosphatase (PTP1B) by Western blot as described previously (17). All primary antibodies were obtained from Cell Signaling Technology, (Danvers, MA) except anti-PTP1B, which was obtained from Abcam (ab2009; Abcam, Cambridge, MA).
Experiment 2: effect of 3rd ventricle low-dose leptin infusions.
The results of experiment 1 indicated that rats receiving low-dose 4th ventricle infusions of leptin gained body fat during the 12 days of leptin infusion. This study was conducted to confirm that low-dose infusions of leptin into the 3rd ventricle produced weight loss. Twenty-eight rats were fitted with a 3rd ventricle cannula as described previously (6). The coordinates for cannula placement on the midline of a level skull relative to the bregma were anteroposterior −2.8 mm, lateral 0.0 mm, and ventral −9.0 mm. Baseline food intakes and body weights were measured for 5 days starting 1 wk after surgery. The rats weighed 320–350 g at the start of the study. At the end of the baseline period, the rats were divided into four weight-matched groups, and an Alzet (model 1002) pump was attached to the cannula delivering PBS or 0.3, 0.6, or 0.9 μg leptin/day. A tail blood sample was collected on day 3 of infusion for measurement of serum leptin. Body composition and serum insulin (rat insulin RIA kit, Millipore), glucose, glycerol (free glycerol reagent F6428; Sigma-Aldrich), free fatty acids (FFA; NEFA C kit, Wako Chemicals, Richmond, VA), and triglyceride (TG) were measured after 12 days of infusion. Liver lipid and glycogen content also were measured as described previously (16).
Experiment 3: effect of 4th ventricle mutein and peripheral leptin infusion.
According to our hypothesis, forebrain and hindbrain leptin receptors have opposing effects on energy balance in the presence of physiological concentrations of leptin. This study tested whether selective blockade of hindbrain receptors with 4th ventricle infusions of leptin mutein protein (mutein; Protein Laboratories Rehovot, Rehovot, Israel) would exaggerate the normal response to a peripheral infusion of leptin. Sixty-four rats were each fitted with a 4th ventricle cannula and allowed to recover from surgery for 1 wk before baseline food intakes and body weights were recorded for 5 days. The rats were divided into eight weight matched groups. Each rat was fitted with an ip Alzet pump (model 2002) that delivered either PBS or 40 μg leptin/day in an infusion volume of 0.5 μl/h. A second Alzet pump (model 1002) was connected to the cannula and delivered saline or 1.5, 2.0, or 3.0 μg mutein/day in an infusion volume of 0.25 μl/h. Daily food intakes and body weights were recorded for a further 12 days, and a tail blood sample was collected on the second day of infusion for measurement of serum leptin concentration. At the end of the study, the rats were euthanized, and body composition was determined. One lobe of the liver was frozen for determination of liver lipid and glycogen content, and serum hormones and metabolites were measured as described above. An additional seven rats per treatment group were fitted with cannulae and Alzet pumps as described above. After 5 days of infusion, two rats per group were anesthetized with ketamine and perfused for immunohistochemical detection of pSTAT3. Blocks of hypothalamic and brain stem tissue were collected from the remaining rats for measurement of pSTAT3 and SOCS3 by Western blot.
Experiment 4: effect of 3rd ventricle mutein and peripheral leptin infusion.
According to our hypothesis, forebrain leptin receptors antagonize the responses produced by hindbrain leptin receptors; therefore, this study tested whether blockade of 3rd ventricle leptin receptors would produce an increase in body fat mass in rats receiving peripheral infusions of leptin. Seventy-two rats were each fitted with a 3rd ventricle cannula and allowed 1 wk to recover from surgery. At the end of 5 days of baseline food intake and body weight measurements, the rats were divided into eight weight-matched groups, and the remainder of the experimental protocol was the same as for experiment 3. An additional two rats per treatment group were fitted with cannulae and Alzet pumps as described above. After 5 days of infusion, the rats were anesthetized with ketamine and perfused for immunohistochemical detection of pSTAT3.
Experiment 5: a dietary model of leptin-induced adiposity.
The studies described above used pharmacological methods to test for differential responses to forebrain and hindbrain leptin. This study tested whether leptin could induce an increase in fat mass in rats that were rapidly gaining body fat and were in the process of becoming leptin resistant. Peripheral infusions of leptin were used, as this is the usual source of leptin that activates central leptin receptors (46). It also was the route of administration that was used when we observed an increase in body fat mass of leptin treated CD rats (18). A pilot study indicated that rats offered ad libitum access to chow, lard, and 30% sucrose solution (choice diet) showed no change in body fat content or food intake if they were offered the choice diet for 4 days before the start of a 12-day peripheral infusion of 40 μg leptin/day. By contrast, the same infusion of leptin reduced body fat mass of rats fed only chow (data not shown). In the present study, the leptin infusion was initiated before the rats were offered the choice diet. Initially all 36 rats in this study were offered chow. Food intakes and body weights were measured for 5 days before they were divided into two weight-matched groups, and each rat was fitted with an ip Alzet pump (model 2002) delivering PBS or 40 μg leptin/day. A small sample of blood was collected by tail bleeding on day 2 for measurement of serum leptin. Starting on day 3 of leptin infusion, one-half of the rats were offered free access to a 30% sucrose solution and lard in addition to chow and water (PBS choice, Leptin choice), and the other half of the rats continued to have free access to chow (PBS chow, Leptin chow). Intake of sucrose, lard, and chow was recorded daily for 7 days, and then the rats were euthanized for determination of body composition and measurement of serum hormones and metabolites. A second cohort of 24 rats was treated as described above, but at the end of the study, sections of hypothalamic and brain stem tissue were collected for measurement of pSTAT3 and SOCS3 by Western blot.
Data analysis.
Statistically significant differences between treatment groups were determined using Statistica software version 9.0 (StatSoft, Tulsa, OK). Differences were considered significant at P < 0.05. Single end–point measurements were compared by unpaired t-test, one-way ANOVA, or two-way ANOVA, depending upon experimental design. Daily measurements of food intake or body weight were compared by repeated-measures ANOVA. Post hoc differences were determined using Duncan's multiple range test. Because we used infusion cannulae for these experiments, it was not possible to test the placement of a third or fourth cannula using behavioral methods. Because carcass fat was our primary end point measurement, rats that had a carcass fat content that was more than 2 standard deviations from the mean for their group were considered outliers and removed from the study.
RESULTS
Experiment 1.
There was no effect of low-dose leptin in the 4th ventricle on food intake, body weight, or weight gain of the rats (Fig. 1). IBAT and core temperature measured by iButtons were averaged over 3 h in the morning (8:30–11:30 AM), when temperatures were at the lowest for the day and 3 h during the evening (7:30–10:30 PM), when temperatures were at their highest on each of the last 9 days of the experiment. The averages for each rat were then averaged for each group. There was no significant effect of leptin on morning IBAT (P < 0.1) or core temperature. IBAT temperature during the dark phase was significantly lower in rats infused with 0.15 μg leptin compared with those infused with 0.6 μg (P < 0.01; Fig. 1). A similar pattern was present in core temperatures, but the difference did not reach significance (P < 0.07). Leptin infusion significantly (P < 0.01) increased carcass fat content by 13% in the 0.6-μg leptin/day group, but the increase in fat content of the 0.15-μg leptin group did not reach significance (P < 0.2; Fig. 1). Although the weight of all fat depots measured tended to be higher in rats infused with 0.6 μg leptin/day than in controls, these differences did not reach significance (data not shown). Serum leptin measured at the end of the study was not different between groups (data not shown).
Fig. 1.

Daily body weight, daily food intake, carcass fat content, core temperature and intrascapular brown fat (IBAT) temperature of rats in experiment 1. Data are means ± SE for groups of 11 or 12 rats receiving 4th ventricle infusions of PBS or leptin for 12 days.
Measurement of leptin signaling proteins in hypothalamic and brain stem tissue after 5 days of infusion of 0.6 μg leptin/day did not show any changes in phosphorylation of STAT3, PI3K, or ERK1/2 in the brain stem (Fig. 2); nor was PTP1B expression increased. SOCS3 was not detected at levels that could be quantified reliably. Surprisingly, infusion of leptin into the 4th ventricle caused a small but significant increase in pPI3K in hypothalamic tissue. Activation of STAT3 in the hypothalamus showed a relatively large variation among individual animals in each treatment group, and although there was a trend for 4th ventricle leptin infusion to increase pSTAT3, this was not statistically significant (Fig. 2). The absence of significant activation of STAT3 in the nucleus of the solitary tract (NTS) and arcuate nucleus of the hypothalamus (ARC) of rats receiving 4th ventricle infusions of leptin was also confirmed by immunohistochemistry (Fig. 3). Because the levels of activation were low in the rats that received 4th ventricle infusions of either PBS or leptin from Alzet pumps for 5 days, tissue from a rat that received an ip injection of 1 mg/kg leptin 30 min before tissue collection is also included in Fig. 3 as a positive control for the sensitivity of the pSTAT3 detection.
Fig. 2.

Western blot analysis of phosphorylation (p) of leptin signaling proteins STAT3, PI3K, and ERK1/2 and expression of SOCS3 and PTP1B in hypothalamic and brain stem tissue of rats receiving 4th ventricle infusions of PBS or 0.6 μg leptin/day for 5 days. Data are means ± SE for 5 rats.
Fig. 3.

Immunohistochemical detection of pSTAT3 in the nucleus of the solitary tract (NTS) and the arcuate nucleus (ARC) of the hypothalamus in rats receiving 4th ventricle infusions of PBS or 0.6 μg/day leptin for 5 days or an ip injection of 1 mg/kg leptin as a positive control. Images of the ARC or NTS are from the same rat for a specific treatment group. AP, area postrema; CC, central canal; SolM, medial nucleus of the solitary tract; 3rd V, 3rd ventricle; ArcM, medial ARC of the hypothalamus; ME, medial eminence. Images were adjusted for contrast and brightness following capture.
Experiment 2.
In this experiment, all rats receiving 3rd ventricle infusions of low doses of leptin lost weight and ate less during the 12 days of the experimental period compared with their controls (Fig. 4). The weight loss was a combination of fat and lean tissue (Fig. 4). Leptin reduced the size of all fat depots weighed, although the retroperitoneal fat from rats infused with 0.6 or 0.9 μg leptin/day was not significantly smaller than that from controls (Table 1). The energy stores of the liver also were significantly depleted, with liver weight, lipid, and glycogen lower in leptin infused than control rats (Table 1). Serum leptin was reduced by ∼50% and serum insulin by as much as 80% in leptin-infused rats compared with controls, but there was no effect of leptin on FFA, TG, or glycerol (data not shown).
Fig. 4.
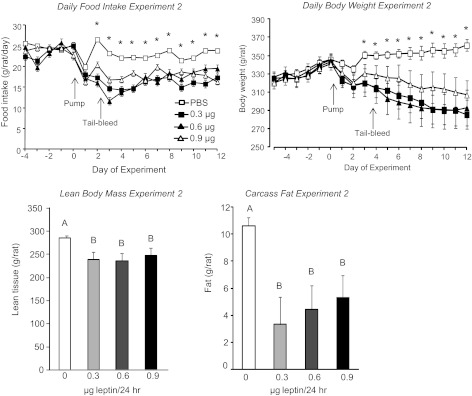
Daily food intake, daily body weight, lean body mass, and carcass fat content of rats that received 3rd ventricle infusions of PBS or leptin for 12 days in experiment 2. Data are means ± SE for groups of 7 rats. *Significant difference in food intake or body weight of PBS- and leptin-infused rats. Values for carcass fat or lean tissue that do not share a common superscript are significantly different at P < 0.05.
Table 1.
Liver composition, fat pad weight, and serum measurements made on rats receiving 3rd ventricle infusions of leptin in experiment 2
| PBS | 0.3 μg Leptin | 0.6 μg Leptin | 0.9 μg Leptin | |
|---|---|---|---|---|
| Liver composition | ||||
| Liver weight, g | 12.3 ± 0.2A | 6.3 ± 1.0B | 7.8 ± 1.0B | 8.4 ± 1.2B |
| Liver lipid, mg | 459 ± 16A | 280 ± 49B | 335 ± 48B | 331 ± 52B |
| Liver glycogen, mg | 220 ± 8A | 137 ± 18B | 145 ± 17B | 167 ± 17B |
| Fat depots | ||||
| Inguinal, g | 3.57 ± 0.19A | 1.38 ± 0.47B | 1.64 ± 0.46B | 2.04 ± 0.54B |
| Epididymal, g | 2.32 ± 0.20A | 0.52 ± 0.35B | 0.84 ± 0.48B | 1.22 ± 0.47B |
| Retroperitoneal, g | 0.65 ± 0.11A | 0.16 ± 0.12B | 0.28 ± 0.16A,B | 0.30 ± 0.12A,B |
| Mesenteric, g | 0.98 ± 0.10A | 0.35 ± 0.12B | 0.39 ± 0.10B | 0.42 ± 0.11B |
| Serum assays | ||||
| Leptin day 3, ng/ml | 2.19 ± 0.18A | 0.96 ± 0.27B | 0.96 ± 0.36B | 1.47 ± 0.42A,B |
| Insulin, ng/ml | 1.02 ± 0.09A | 0.19 ± 0.09B | 0.27 ± 0.12B | 0.49 ± 0.28B |
| Glucose, mg/dl | 129 ± 10A | 99 ± 13B | 111 ± 13B | 96 ± 7B |
Data are means ± SE for groups of 7 rats. Fat pad weight and all serum measurements except leptin were recorded after 12 days of leptin infusion into the 3rd ventricle. Values for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Experiment 3.
In this experiment, rats received peripheral infusions of 40 μg leptin/day and increasing doses of mutein into the 4th ventricle. Food intake was inhibited in all leptin-infused rats, but central mutein infusion had no effect on food intake (Fig. 5: leptin, P < 0.001; mutein, NS; interaction, NS). Similarly, leptin inhibited weight gain of the rats (leptin, P < 0.001; mutein, NS; interaction, NS), with the greatest effect in leptin-treated rats receiving 3.0 μg mutein/24 h (Table 2). At the end of the experiment, the weights of inguinal and retroperitoneal fat depots were smaller in leptin-infused than in PBS-infused rats (leptin, P < 0.001; mutein, NS; interaction, NS), but there was no effect of leptin or mutein on the weight of mesenteric or epididymal fat (Table 2). Carcass fat content of all leptin-infused rats was lower than that of the PBS-infused rats that were not infused with mutein. Fourth ventricle mutein infusions caused a dose-related inhibition of carcass fat such that the PBS-infused rats receiving 3.0 μg mutein had the same amount of fat as rats receiving peripheral leptin infusions and significantly less fat than PBS-infused rats not receiving mutein (Fig. 5: leptin, P < 0.001; mutein, P < 0.07; interaction, NS). There was no independent effect of leptin on lean tissue, but mutein infusions significantly reduced lean body mass of the rats (Fig. 5: leptin, NS; mutein, P < 0.01; interaction, NS). PBS-infused rats receiving 3.0 μg mutein/day had significantly less lean tissue than those infused with 0 or 1.5 μg mutein/day. Leptin-infused rats receiving 3.0 μg mutein/day had significantly less lean tissue than PBS-infused rats receiving 0 μg mutein, whereas there was no difference in lean mass of leptin-infused and PBS-infused rats receiving 0 μg mutein/day. Despite these changes in carcass composition, there was no effect of either leptin or mutein on liver weight, lipid, or glycogen content (data not shown). There was no effect of leptin or mutein on serum FFA, TG, or glycerol concentrations (data not shown). Leptin tended to reduce blood glucose, and this was reversed by mutein infusion (Table 2: leptin, NS; mutein, P < 0.04; interaction, NS) without any significant effect on insulin.
Fig. 5.

Total food intake, carcass fat, and lean body mass of rats in experiment 3 that received peripheral infusions of PBS or 40 μg leptin/day and 4th ventricle infusions of increasing doses of mutein for 12 days. Data are means ± SE for groups of 8 rats. Values for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Table 2.
Weight gain, fat pad weight, and serum measurments made on rats receiving 4th ventricle infusions of mutein and peripheral infusion of leptin in experiment 3
| PBS |
Leptin |
|||||||
|---|---|---|---|---|---|---|---|---|
| Saline | 1.5 μg Mutein | 2.0 μg Mutein | 3.0 μg Mutein | Saline | 1.5 μg Mutein | 2.0 μg Mutein | 3.0 μg Mutein | |
| Start wt, g | 365 ± 5 | 371 ± 5 | 366 ± 4 | 360 ± 7 | 360 ± 6 | 361 ± 5 | 358 ± 4 | 362 ± 1 |
| Wt gain, g | 21 ± 3A | 17 ± 2A,C | 16 ± 4A,C | 17 ± 3A,C | 12 ± 3B,C | 11 ± 2B,C | 10 ± 4B,C | 3 ± 5B |
| Fat depots | ||||||||
| Inguinal, g | 4.8 ± 0.2A | 4.8 ± 0.3A | 4.4 ± 0.3A,B | 4.3 ± 0.2A,B | 3.8 ± 0.2B | 3.7 ± 0.3B | 3.6 ± 0.2B | 4.1 ± 0.4B |
| Epididymal, g | 3.3 ± 0.2 | 3.3 ± 0.2 | 2.9 ± 0.1 | 3.7 ± 0.2 | 3.3 ± 0.2 | 3.4 ± 0.2 | 3.2 ± 0.1 | 3.7 ± 0.2 |
| Retroperitoneal, g | 1.2 ± 0.1A | 1.2 ± 0.1A | 1.0 ± 0.1A,B | 1.0 ± 0.1A,B | 0.7 ± 0.1B,C | 0.7 ± 0.9B,C | 0.5 ± 0.1C | 0.9 ± 0.2B,C |
| Mesenteric, g | 1.7 ± 0.1 | 1.6 ± 0.1 | 1.6 ± 0.2 | 1.8 ± 0.1 | 1.9 ± 0.1 | 1.9 ± 0.1 | 2.0 ± 0.1 | 1.8 ± 0.2 |
| Serum assays | ||||||||
| Leptin day 3, ng/ml | 3.6 ± 0.6A | 5.0 ± 0.6A,B | 4.8 ± 0.6A,B | 4.4 ± 0.6A,B | 6.0 ± 0.6B | 4.4 ± 0.4A,B | 4.6 ± 0.6A,B | 5.4 ± 0.8A,B |
| Insulin, ng/ml | 1.04 ± 0.10 | 1.00 ± 0.12 | 0.93 ± 0.09 | 1.00 ± 0.24 | 0.71 ± 0.10 | 0.86 ± 0.14 | 1.09 ± 0.18 | 0.76 ± 0.11 |
| Glucose, mg/dl | 129 ± 10AB | 137 ± 3AB | 120 ± 5A | 139 ± 11A,B | 116 ± 4A | 139 ± 5A,B | 137 ± 11A,B | 150 ± 6B |
Data are means ± SE for groups of 8 rats receiving 4th ventricle infusions of mutein and peripheral infusions of PBS or leptin for 12 days. Measurements of tissue weights and serum hormones, other than leptin, were made at the end of the study. Values for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Western blots showed that peripheral leptin infusion significantly increased pSTAT3 in the brain stem but not the hypothalamus of rats that received 4th ventricle infusions of saline (Fig. 6). Fourth ventricle mutein tended to increase pSTAT3 in both the hypothalamus and brain stem of all rats, but this was not significant (P < 0.1). Immunohistochemistry did not illustrate any dramatic effect of leptin or mutein infusion on pSTAT3 in either the ARC or the NTS of the rats (Fig. 6). The increase in brain stem pSTAT3 appeared to be associated with activation of the area postrema rather than the NTS.
Fig. 6.

Hypothalamic and brain stem pSTAT3 measured by Western blot (top, n = 5) for rats that received peripheral infusions of PBS or 40 μg leptin/day and 4th ventricle infusions of saline or 3.0 μg mutein/24 h for 5 days in experiment 3. Bottom: representative immunohistochemical detection of pSTAT3 in the ARC and NTS of rats that received peripheral infusions of PBS or 40 μg leptin/day and 4th ventricle infusions of saline or 3.0 μg mutein/24 h mutein for 5 days. Images were adjusted for contrast and brightness following capture.
Experiment 4.
In this experiment, rats received peripheral infusions of 40 μg leptin/day and increasing doses of mutein into the 3rd ventricle. The objective of the experiment was to determine whether peripheral infusions of leptin in the absence of activation of forebrain leptin receptors would increase body fat mass. Leptin infusion caused a nonsignificant reduction in food intake of the rats, and this was reversed by 3rd ventricle infusion of mutein. Leptin-infused rats receiving 3.0 μg mutein/day had a significantly higher food intake than those receiving 3rd ventricle infusions of saline but the same intake as PBS-infused rats receiving 3.0 μg mutein/day (Fig. 7: leptin, NS; mutein, P < 0.02; interaction, NS). There was no significant effect of leptin or mutein on weight gain of the rats (Table 3), but at the end of the experiment, leptin-treated rats infused with 3.0 μg mutein/day were fatter than those infused with saline (Fig. 7: leptin, NS; mutein, P < 0.02; interaction, NS). They had the same body fat mass as PBS-infused rats receiving 3.0 μg mutein/day. The increase in body fat was associated with enlargement of inguinal and retroperitoneal fat pads, but there was no change in epididymal or mesenteric fat (Table 3). There was no effect of leptin or mutein on lean body mass (Fig. 7), liver weight, lipid, or glycogen content (data not shown). Serum leptin was increased by central mutein infusion, and this effect was exaggerated by peripheral infusions of leptin (Table 3: leptin, P < 0.001; mutein, P < 0.05; interaction, NS). Serum insulin was reduced by peripheral leptin infusion, but this was reversed with central mutein infusion (Table 3: leptin, NS; mutein, NS; interaction, P < 0.05). Similar to the previous experiments, the low-dose leptin infusion had a minimal effect on pSTAT3 in the ARC and the NTS detected by immunohistochemistry. The combination of peripheral leptin and 3rd ventricle mutein did increase hypothalamic pSTAT3, but the activation remained much less intense than was produced by ip injection of 1 mg/kg leptin (see Figs. 8 and 3).
Fig. 7.

Total food intake, carcass fat, and lean body mass of rats in experiment 4 that received peripheral infusions of PBS or 40 μg leptin/day and 3rd ventricle infusions of increasing doses of mutein for 12 days. Data are means ± SE for groups of 9 rats. Values for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Table 3.
Weight gain, fat pad weight, and serum measurments made on rats receiving 3rd ventricle infusions of mutein and peripheral infusion of leptin in experiment 4
| PBS |
Leptin |
|||||||
|---|---|---|---|---|---|---|---|---|
| Saline | 1.5 μg Mutein | 2.0 μg Mutein | 3.0 μg Mutein | Saline | 1.5 μg Mutein | 2.0 μg Mutein | 3.0 μg Mutein | |
| Start wt, g | 347 ± 6 | 344 ± 5 | 344 ± 5 | 343 ± 5 | 343 ± 7 | 345 ± 4 | 348 ± 6 | 351 ± 6 |
| d | 10 ± 3 | 15 ± 2 | 14 ± 7 | 20 ± 3 | 5 ± 4 | 9 ± 3 | 18 ± 5 | 16 ± 5 |
| Fat Depots | ||||||||
| Inguinal, g | 3.8 ± 0.4A,B | 3.8 ± 0.2A,B | 3.7 ± 0.4A,B | 4.2 ± 0.2B | 3.0 ± 0.2A | 3.4 ± 0.3A,B | 3.5 ± 0.3A,B | 4.1 ± 0.2B |
| Epididymal, g | 3.5 ± 0.3 | 3.3 ± −0.3 | 3.3 ± 0.3 | 3.4 ± 0.2 | 3.1 ± 0.2 | 3.4 ± 0.1 | 3.0 ± 0.3 | 3.8 ± 0.2 |
| Retroperitoneal, g | 0.8 ± 0.1A,B | 0.8 ± 0.1A,B | 0.8 ± 0.1A,B | 1.1 ± 0.1B | 0.6 ± 0.1A | 0.6 ± 0.1A | 0.7 ± 0.1A,B | 1.0 ± 0.1B |
| Mesenteric, g | 2.3 ± 0.3 | 2.0 ± 0.2 | 2.0 ± 0.1 | 2.3 ± 0.2 | 1.7 ± 0.2 | 1.8 ± 0.1 | 1.9 ± 0.2 | 1.9 ± 0.1 |
| Serum assays | ||||||||
| Leptin day 2, ng/ml | 2.0 ± 0.5A | 3.0 ± 0.6AC | 3.3 ± 0.7A,C | 3.6 ± 0.5A,C | 4.0 ± 1.0A,C | 5.5 ± 0.7B,C | 7.9 ± 1.6B | 4.5 ± 0.8A,C |
| Insulin, ng/ml | 1.0 ± 0.2A | 0.8 ± 0.1A,B | 1.0 ± 0.1A | 1.2 ± 0.1A | 0.7 ± 0.1B | 1.0 ± 0.2A | 1.1 ± 0.1A | 0.9 ± 0.1A,B |
| Glucose, mg/dl | 155 ± 13A | 139 ± 8A,B | 136 ± 13A,B | 114 ± 8A | 125 ± 7A,B | 152 ± 12B | 119 ± 8A | 139 ± 12A,B |
Data are means ± SE for groups of 9 rats receiving 3rd ventricle infusions of mutein and peripheral infusions of PBS or leptin for 12 days. Measurements of tissue weights and serum hormones, other than leptin, were made at the end of the study. Values for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Fig. 8.
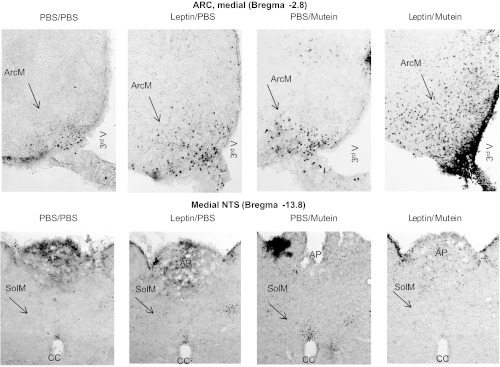
Representative immunohistochemical detection of pSTAT3 in the ArcM and medial NTS of rats in experiment 4 that received peripheral infusions of PBS or 40 μg leptin/day and 3rd ventricle infusions of 3.0 μg mutein/24 h for 5 days. Images were adjusted for contrast and brightness following capture.
Experiment 5.
This experiment was designed to test whether the leptin-induced gain in body fat that was achieved by pharmacological means in experiment 1 could be extended to rats that were developing diet-induced leptin resistance. The peripheral infusions of leptin caused a doubling of circulating leptin concentrations measured on day 2 of infusion (Table 4: infusion, P < 0.01; diet, NS; interaction, NS). The difference between PBS- and leptin-infused chow rats had disappeared by day 10 of the experiment, possibly due to a downregulation of endogenous leptin, but was still present in the choice rats (Table 4: infusion, P < 0.001; diet, P < 0.001; interaction, P < 0.01). Energy intake was significantly increased in choice rats (Fig. 9: infusion, NS; diet, P < 0.01; interaction, NS), with ∼15% of energy intake contributed by sucrose solution, 45–50% by lard, and 35–40% by chow (Fig. 9). There was no effect of leptin on the energy intake of either chow or choice rats. At the end of the experiment, choice-fed rats were significantly fatter than their chow-fed controls, with an increase in the size of all fat pads measured and in liver lipid content (Table 4: infusion, NS; diet, P < 0.01; interaction, NS). Leptin caused a nonsignificant decrease in carcass fat content of chow-fed rats but a significant increase in the choice-fed rats (Fig. 9: infusion, NS; diet, P < 0.04; interaction, P < 0.01). There was no significant effect of diet or leptin on lean body mass of the rats (Fig. 9). Choice diet caused a mild hyperinsulinemia, and leptin-infused chow rats had lower glucose levels than choice rats (Table 4). Western blot analysis did not show any significant effect of diet or peripheral leptin infusion on hypothalamic pSTAT3 or SOCS3 (Fig. 10). In the brain stem, there was no effect of diet or leptin on pSTAT3. SOCS3 was increased by the choice diet (P < 0.03), but post hoc analysis did not show significant differences between specific groups.
Table 4.
Liver composition, fat pad weight,and serum measurments made on rats receiving peripheral infusions of leptin in experiment 5
| PBS Chow | Leptin Chow | PBS Choice | Leptin Choice | |
|---|---|---|---|---|
| Liver weight, g | 10.5 ± 0.2 | 10.6 ± 0.3 | 11.1 ± 0.4 | 10.8 ± 0.4 |
| Liver lipid, mg | 0.43 ± 0.01A | 0.41 ± 0.01A | 0.48 ± 0.02B | 0.48 ± 0.03B |
| Fat depots | ||||
| Inguinal, g | 2.62 ± 0.09A | 2.24 ± 0.15A | 3.32 ± 0.22B | 3.47 ± 0.20B |
| Epididymal, g | 1.51 ± 0.09A | 1.47 ± 0.05A | 1.74 ± 0.07A,B | 1.95 ± 0.15B |
| Retroperitoneal, g | 0.67 ± 0.07A | 0.52 ± 0.06A | 0.95 ± 0.08B | 1.04 ± 0.12B |
| Mesenteric, g | 1.87 ± 0.1A | 1.67 ± 0.18A | 2.48 ± 0.14B | 2.68 ± 0.21B |
| Serum assays | ||||
| Leptin day 2, ng/ml | 3.4 ± 0.4A | 7.7 ± 1.5B | 4.0 ± 0.5A | 7.6 ± 1.1B |
| Leptin day 10, ng/ml | 2.4 ± 0.1A | 2.9 ± 0.1A | 2.7 ± 0.2A | 5.9 ± 1.0B |
| Insulin, ng/ml | 0.42 ± 0.02A | 0.35 ± 0.04A | 0.53 ± 0.06A,B | 0.61 ± 0.06B |
| Glucose, mg/dl | 137 ± 4A,B | 126 ± 5B | 148 ± 7A | 150 ± 4A |
| Triglyceride, mg/dl | 24 ± 2 | 27 ± 2 | 30 ± 3 | 30 ± 2 |
Data are means ± SE for groups of 9 rats. Leptin-treated rats were infused with 40 μg leptin/24 h for 10 days, and choice rats received choice diet for the last 7 days of infusion. Values for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Fig. 9.

Total energy intake during the 3 days when rats were infused with leptin, but all groups were offered chow. Total energy intake during the 7 days when rats were infused with leptin and offered either chow or choice diet. Daily energy intake during the the experiment. Carcass fat and lean body mass of rats in experiment 5 that were fed chow or choice diet ad libitum for the last 7 days of a 10-day peripheral infusion of PBS or 40 μg leptin/day. Data for a specific parameter that do not share a common superscript are significantly different at P < 0.05.
Fig. 10.

Western blot analysis of pSTAT3 and SOCS3 in tissue blocks of the hypothalamus and brain stem of rats in experiment 5 that were fed chow or choice diet ad libitum for the last 7 days of a 10-day peripheral infusion of PBS or 40 μg leptin/day. Data are means ± SE for groups of 6 rats.
DISCUSSION
The primary objective of the studies described here was to test whether selective activation of leptin receptors in the hindbrain would increase body fat mass and replicate the change in body composition that we had previously found in CD rats that received peripheral infusions of leptin (18). A secondary objective was to determine whether selective blockade of leptin receptors available to the 3rd or 4th ventricle would modify the whole animal response to peripherally administered leptin. Experiment 1 indicated that leptin infusions in the 4th ventricle could produce a small but significant increase in body fat mass of rats over 12 days. Lean body mass was not measured in these rats; however, in experiment 5 leptin increased carcass fat content of rats fed the choice diet with no effect on lean body mass, and it is likely that 4th ventricle leptin acted specifically to increase body fat. A specific effect on fat would explain why we found no significant differences in weight gain of the rats, because the 13% (4 g) increase in fat would not be detected as a change in weight of rats that were 430 g at the end of the study. The increase in body fat occurred without a measurable increase in daily food intake, suggesting an increase in efficiency of energy utilization. The assumption that energy expenditure was reduced would be consistent with the leptin-induced increase in carcass fat of CD rats being associated with an inhibition of energy expenditure during the light period (18). The small decrease in IBAT temperature of rats infused with 0.15 μg leptin in experiment 1 supports the notion that thermogenesis was inhibited by extremely low doses of leptin in the hindbrain, but this effect was not apparent in the rats receiving 4th ventricle infusions of 0.6 μg leptin/day even though their carcass fat content was increased. Therefore, if the change in body composition of leptin-treated rats was dependent on a decrease in daily expenditure, then it would have to be due to a change in activity or resting expenditure, neither of which were measured in experiment 1.
In a previous study, we reported that high-fat-fed mice receiving peripheral infusions of leptin and housed in a warm environment (27°C) gained body fat mass when compared with their PBS-infused controls (19). We hypothesized that the increase in carcass fat was due to a partial leptin resistance that prevented leptin from reducing food intake and body fat combined with housing in an environment that prevented an increase in energy expenditure. The results of the studies described here suggest that leptin increases body fat when there is selective activation of receptors located in the hindbrain. These observations would be consistent with a report by Munzberg et al. (29), who found that diet-induced leptin resistance was associated with a failure to respond to leptin in only selected areas of the brain in mice. In addition, Levin et al. (25) reported that leptin-induced activation of STAT3 in the NTS, but not the hypothalamus, of diet-induced obese rats, which may have been similar to the situation for rats in experiment 5, in which leptin increased body fat of rats offered a choice diet during the period of leptin infusion.
Because the selective activation of 4th ventricle leptin receptors is anabolic and increases body fat, whereas 3rd ventricle leptin decreases food intake and causes tissue loss, experiments 3 and 4 tested whether these two areas modulated the leptin response in an animal receiving peripheral infusions of leptin. The results clearly show that in nonstimulated conditions 4th ventricle leptin receptors have an anabolic effect, but they do not attenuate the whole animal response to peripheral leptin infusions. If 3rd and 4th ventricle leptin receptors always acted in opposition to each other, then in experiment 4 we should have found that blockade of 3rd ventricle leptin receptors exaggerated weight gain in leptin infused rats. By contrast, although the highest dose of mutein did cause weight gain in the leptin-infused rats, this was no different from the response in PBS-infused rats. These results suggest that the anabolic pathway is easily overridden by activation of leptin receptors in sites outside the 4th ventricle. This appears to be the case even when the changes in leptin concentrations are relatively small, because the infusions used in experiments 3 and 4 only doubled serum leptin concentration compared with control rats. The hindbrain leptin receptors do, however, protect lean body mass even in leptin-stimulated conditions, because 4th ventricle mutein infusions reduced lean body mass in control rats and exaggerated the loss of lean tissue in leptin-treated animals in experiment 3. It is worth noting that the leptin-induced loss of lean mass was significant only when leptin was given centrally (experiment 2) or when the activity of receptors in the region of the 3rd ventricle receptors was increased relative to that of receptors in the vicinity of the 4th ventricle (experiment 3), because there were no significant changes in lean body mass of rats receiving peripheral infusions of leptin with no modification of central leptin signaling (no mutein rats in experiments 3 and controls in experiment 5) or with blockade of 3rd ventricle leptin receptors (experiment 4).
Hayes et al. (21) have reported that knockdown of leptin receptors in the NTS and area postrema increases food intake and body fat content of rats. Data from experiment 1 would obviously contradict the conclusion that leptin receptors in the hindbrain prevent hyperphagia; however, in the study by Hayes et al., which reduced leptin receptor expression by 40%, chow-fed rats lost weight during the first 9 days after receptor knockdown, and there was no effect on food intake during the first 2 wk of the study, which is equivalent to the time course of our experiment. The increase in food intake of rats with leptin receptor knockdown was apparent only in weeks 3 and 5 after treatment when intake was averaged over the week. Similarly, average weekly weight gain was not different until 4 wk after receptor knockdown. There was a more reliable hyperphagia and exaggerated weight gain when the rats were offered a 60% kcal fat diet and the body fat content of the receptor knockdown rats was increased compared with controls after 4 wk on the high-fat diet. Therefore, although the results of the experiments described here would appear to be the opposite of those expected from the leptin receptor knockdown study, the difference in timing of measurements may explain the discrepant results. Body composition was not measured at an early time point by Hayes et al., but the 9-day weight loss in the receptor knockdown rats may have been similar to the loss of fat and lean tissue in rats in experiment 3, when 4th ventricle leptin receptors were blocked by mutein infusion. Comparison of these two studies raises the question of whether we would find an adaptation to the inhibition of 4th ventricle leptin receptors if we extended our infusion studies beyond 12 days and whether adiposity would increase if the rats were fed a high-fat diet instead of chow.
The increase in body fat mass of rats receiving 4th ventricle infusions of leptin is contrary to reports of leptin-induced hypophagia and weight loss in rats receiving 4th ventricle leptin injections (23, 36, 41). There are a number of differences between the studies that report hypophagia and experiment 1 described here. The first is the method of leptin administration (chronic infusions compared with single acute injections), and the dose of leptin used was much higher in the studies showing an inhibition of food intake (3–10 μg in a single injection vs. 0.6 μg over 24 h). The study by Skibicka et al. (41) indicated that a high dose was required to produce an effect on feeding because a single 4th ventricle injection of 3 μg of leptin did not significantly inhibit food intake or weight gain of the rats. Similarly, Huo et al. (23) showed that 5 μg of leptin was subthreshold in that it inhibited food intake only if the rats also experienced gastric distention, illustrating the importance of the integration of central and peripheral signals in determining food consumption.
We were not able to identify the signaling mechanisms responsible for weight gain in rats receiving 4th ventricle infusions of leptin. Western blot and immunohistochemsitry showed no significant activation of STAT3 in either the hypothalamus or brain stem; in addition, there was no obvious activation of PTP1B or SOCS3, both of which are proteins that inhibit leptin signaling (2, 3). In experiment 1, we did find that 4th ventricle leptin increased phosphorylation of PI3K in the hypothalamus, but this was not associated with any change in food intake or body weight; therefore, the physiological function of this activation needs further investigation. By contrast to our results, Ruiter et al. (36) reported that an injection of leptin into the 4th ventricle that caused weight loss also increased pSTAT3 in hypothalamic nuclei. They suggested that, if leptin was activating receptors in the hypothalamus, then it was possible that the 4th ventricle leptin was being carried to the hypothalamus in the subarachanoid space. This mechanism is unlikely to explain their additional observations that parenchymal injection of 50 ng of leptin directly into the NTS also increased phosphorylation of STAT3 in the hypothalamus. Because activation of leptin receptors is not the exclusive stimulus for phosphorylation of STAT3, it is possible that leptin stimulated neurons that project from the NTS to the forebrain and that these neurons released or inhibited other neurotransmitters that regulate STAT3 phosphorylation. It has been established in other systems that cytokines and growth factors (8), serotonin (31), and glutamate (30) stimulate STAT3 phosphorylation, whereas dopamine inhibits STAT3 phosphorylation acting through the D2R receptor (24). Therefore, further studies are also required to determine how leptin in the hindbrain activates STAT3 in the forebrain. In the experiments described here, low-dose leptin infusions into the 4th ventricle did not stimulate pSTAT3 in the hypothalamus, but it is possible that if we had used higher doses we would have seen an increase in hypothalamic pSTAT3 and weight loss in the rats. Because we did not detect significant changes in central pSTAT3 in rats that showed significant changes in body composition it is possible that the leptin was acting through a different signaling cascade, although it also is possible that small changes in STAT3 activation are enough to change energy balance.
By contrast to the effects of low-dose leptin infusions into the 4th ventricle, equivalent doses of leptin into the 3rd ventricle produced a dramatic inhibition of food intake and body fat content. The lowest dose tested here (0.3 μg/24 h) produced a maximal effect on these parameters, indicating that the hypothalamic leptin receptors have a lower threshold for response than those located around the 4th ventricle. These results are entirely consistent with those in the literature but were achieved using lower doses of leptin than have been used by others investigating the effects of central leptin infusions on energy balance. It is of interest that when leptin was infused into the 3rd ventricle the loss of fat was associated with a change in weight of all of the fat depots measured. By contrast, when leptin was given peripherally, the gain or loss of fat in rats receiving central infusions of mutein were associated with changes in weight of only the inguinal and retroperitoneal depots, with no change in the mesenteric or epididymal depots. It is possible that leptin influences the size of different fat depots through different mechanisms and that the inguinal and retroperitoneal depots are more dependent on central control. We (34) have previously reported differential changes in norepinephrine turnover in the response of individual white fat depots to peripheral or 3rd ventricle leptin. In addition, leptin receptors are expressed in many areas of the rat brain other than the hypothalamus or brain stem (11, 13) as well as in peripheral tissues (10), including adipocytes (28); thus, there are multiple potential sites at which leptin could regulate the size of mesenteric and epididymal fat that would not have been disrupted by 3rd or 4th ventricle infusions of mutein.
We did not find any significant changes in serum FFA, glycerol, or TG concentrations in any of the experiments described here, including experiment 2, where 3rd ventricle infusions of leptin caused a substantial reduction in body fat mass and liver lipid content. By contrast, both 3rd ventricle leptin infusions in experiment 2 and peripheral leptin infusions in experiments 3 and 4 reduced blood glucose and serum insulin concentrations, consistent with previous reports that peripheral or central leptin administration increases insulin sensitivity (35, 39, 40). The low-dose peripheral infusions of leptin used in experiments 3, 4, and 5 caused a doubling of serum leptin concentrations, suggesting that we were within a physiological range of response. Previously, we and others (26, 32, 34) have reported that single central injections of leptin increased circulating concentrations of leptin, and this did not occur with the low-dose leptin infusions used in experiment 2. Surprisingly, 4th ventricle infusions of mutein increased circulating leptin concentrations even though body fat mass was reduced. These results suggest that the increase in circulating leptin is caused by a relative increase in activity of forebrain leptin receptors, either due to direct application of leptin, or because of a downregulation of the activity of leptin receptors adjacent to the 4th ventricle.
In summary, the experiments described here show that selective activation of leptin receptors in the hindbrain with low doses of leptin causes a small but significant increase in body fat. This response can be reproduced in animals that are in the early stages of development of diet-induced leptin resistance. The basal activity of leptin receptors in the hindbrain may play a protective role, preventing the catabolism of lean and fat tissue when leptin concentrations in the hypothalamus are at nonstimulated levels. The leptin-induced increase in adipose tissue may be dependent on a disruption of the balance between forebrain and hindbrain leptin receptors, as would occur with the site-selective development of leptin resistance in diet-induced obesity. The unexpected anabolic action of hindbrain leptin receptors does not appear to be in play when leptin is infused peripherally, suggesting that the anabolic action of these receptors is easily overcome by activation of leptin receptors in other central and/or peripheral tissues. The difference in results reported here with reports from studies that show of weight loss in rats that received 4th ventricle injections of leptin (23, 41) may be due to the higher doses of leptin being used for the injections in the hindbrain leading to a simultaneous activation of forebrain leptin receptors (36).
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-053903 awarded to R. B. S. Harris.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.B.H. conception and design of research; R.B.H. performed experiments; R.B.H. analyzed data; R.B.H. interpreted results of experiments; R.B.H. prepared figures; R.B.H. drafted manuscript; R.B.H. edited and revised manuscript; R.B.H. approved final version of manuscript.
ACKNOWLEDGMENTS
I thank Mary Shaw for performing the Western blots in experiment 1.
REFERENCES
- 1. Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides 17: 305–311, 1996 [DOI] [PubMed] [Google Scholar]
- 2. Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12: 917–924, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS3as a potential mediator of central leptin resistance. Mol Cell 1: 619–625, 1998 [DOI] [PubMed] [Google Scholar]
- 4. Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, Su K, Cheng B, Li X, Harvey-White J, Schwartz GJ, Kunos G, Rossetti L. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med 14: 667–675, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi YH, Li C, Hartzell DL, Little DE, Della-Fera MA, Baile CA. Icv leptin effects on spontaneous physical activity and feeding behavior in rats. Behav Brain Res 188: 100–108, 2008 [DOI] [PubMed] [Google Scholar]
- 6. Chotiwat C, Harris RB. Antagonism of specific corticotropin-releasing factor receptor subtypes selectively modifies weight loss in restrained rats. Am J Physiol Regul Integr Comp Physiol 295: R1762–R1773, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. New Eng J Med 334: 292–295, 1996 [DOI] [PubMed] [Google Scholar]
- 8. Devarajan E, Huang S. Stat3 as a central regulator of tumor metastases. Curr Mol Med 9: 626–633, 2009 [DOI] [PubMed] [Google Scholar]
- 9. El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 105: 1827–1832, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fruhbeck G. Peripheral actions of leptin, and its involvement in disease. Nutr Rev 60: S47–S55; discussion S68-S84, 85–47, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Guan XM, Hess JF, Yu H, Hey PJ, van der Ploeg LH. Differential expression of mrna for leptin receptor isoforms in the rat brain. Mol Cell Endocrinol 133: 1–7, 1997 [DOI] [PubMed] [Google Scholar]
- 12. Gullicksen PS, Flatt WP, Dean RG, Hartzell DL, Baile CA. Energy metabolism and expression of uncoupling proteins 1, 2, and 3 after 21 days of recovery from intracerebroventricular mouse leptin in rats. Physiol Behav 75: 473–482, 2002 [DOI] [PubMed] [Google Scholar]
- 13. Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci 18: 559–572, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haring SJ, Harris RB. The relation between dietary fructose, dietary fat and leptin responsiveness in rats. Physiol Behav 104: 914–922, 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harris RB. Growth measurements in sprague-dawley rats fed diets of very low fat concentration. J Nutr 121: 1075–1080, 1991 [DOI] [PubMed] [Google Scholar]
- 16. Harris RB. Factors influencing energy intake of rats fed either a high-fat or a fat mimetic diet. Int J Obes Relat Metab Disord 18: 632–640, 1994 [PubMed] [Google Scholar]
- 17. Harris RB, Apolzan JW. Changes in glucose tolerance and leptin responsiveness of rats offered a choice of lard, sucrose and chow. Am J Physiol Regul Integr Comp Physiol 302: R1327–R1339, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harris RB, Bartness TJ, Grill HJ. Leptin responsiveness in chronically decerebrate rats. Endocrinology 148: 4623–4633, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Harris RB, Mitchell TD, Kelso EW, Flatt WP. Changes in environmental temperature influence leptin responsiveness in low- and high-fat-fed mice. Am J Physiol Regul Integr Comp Physiol 293: R106–R115, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Harris RB, Zhou J, Redmann SM, Jr., Smagin GN, Smith SR, Rodgers E, Zachwieja JJ: A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology 139: 8–19, 1998 [DOI] [PubMed] [Google Scholar]
- 21. Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, Grill HJ. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 11: 77–83, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horn CC, Kaplan JM, Grill HJ, Friedman MI. Brain fos-like immunoreactivity in chronic decerebrate and neurologically intact rats given 2,5-anhydro-d-mannitol. Brain Res 801: 107–115, 1998 [DOI] [PubMed] [Google Scholar]
- 23. Huo L, Maeng L, Bjorbaek C, Grill HJ. Leptin and the control of food intake: Neurons in the nucleus of the solitary tract are activated by both gastric distension and leptin. Endocrinology 148: 2189–2197, 2007 [DOI] [PubMed] [Google Scholar]
- 24. Kim KS, Yoon YR, Lee HJ, Yoon S, Kim SY, Shin SW, An JJ, Kim MS, Choi SY, Sun W, Baik JH. Enhanced hypothalamic leptin signaling in mice lacking dopamine d2 receptors. J Biol Chem 285: 8905–8917, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol 286: R143–R150, 2004 [DOI] [PubMed] [Google Scholar]
- 26. Maness LM, Kastin AJ, Farrell CL, Banks WA. Fate of leptin after intracerebroventricular injection into the mouse brain. Endocrinology 139: 4556–4562, 1998 [DOI] [PubMed] [Google Scholar]
- 27. Miragaya JR, Harris RB. Antagonism of corticotrophin-releasing factor receptors in the fourth ventricle modifies responses to mild but not restraint stress. Am J Physiol Regul Integr Comp Physiol 295: R404–R416, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muller G, Ertl J, Gerl M, Preibisch G. Leptin impairs metabolic actions of insulin in isolated rat adipocytes. J Biol Chem 272: 10585–10593, 1997 [DOI] [PubMed] [Google Scholar]
- 29. Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 145: 4880–4889, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, Collingridge GL: The JAK/STAT. pathway is involved in synaptic plasticity. Neuron 73: 374–390, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oufkir T, Vaillancourt C. Phosphorylation of JAK2 by serotonin (2A)-HT receptor activates both STAT3 and ERK1/2 pathways and increases growth of JEG-3 human placental choriocarcinoma cell. Placenta 32: 1033–1040, 2011 [DOI] [PubMed] [Google Scholar]
- 32. Pal R, Sahu A. Leptin signaling in the hypothalamus during chronic central leptin infusion. Endocrinology 144: 3789–3798, 2003 [DOI] [PubMed] [Google Scholar]
- 33. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269: 540–543, 1995 [DOI] [PubMed] [Google Scholar]
- 34. Penn DM, Jordan LC, Kelso EW, Davenport JE, Harris RB. Effects of central or peripheral leptin administration on norepinephrine turnover in defined fat depots. Am J Physiol Regul Integr Comp Physiol 291: R1613–R1621, 2006 [DOI] [PubMed] [Google Scholar]
- 35. Pocai A, Morgan K, Buettner C, Gutierrez-Juarez R, Obici S, Rossetti L. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes 54: 3182–3189, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Ruiter M, Duffy P, Simasko S, Ritter RC. Increased hypothalamic signal transducer and activator of transcription 3 phosphorylation after hindbrain leptin injection. Endocrinology 151: 1509–1519, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scarpace PJ, Matheny M, Pollock BH, Tumer N. Leptin increases uncoupling protein expression and energy expenditure. Am J Physiol Endocrinol Metab 273: E226–E230, 1997 [DOI] [PubMed] [Google Scholar]
- 38. Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience 104: 1111–1117, 2001 [DOI] [PubMed] [Google Scholar]
- 39. Shi ZQ, Nelson A, Whitcomb L, Wang J, Cohen AM. Intracerebroventricular administration of leptin markedly enhances insulin sensitivity and systemic glucose utilization in conscious rats. Metab Clin Exper 47: 1274–1280, 1998 [DOI] [PubMed] [Google Scholar]
- 40. Sivitz WI, Walsh SA, Morgan DA, Thomas MJ, Haynes WG. Effects of leptin on insulin sensitivity in normal rats. Endocrinology 138: 3395–3401, 1997 [DOI] [PubMed] [Google Scholar]
- 41. Skibicka KP, Grill HJ. Hindbrain leptin stimulation induces anorexia and hyperthermia mediated by hindbrain melanocortin receptors. Endocrinology 150: 1705–1711, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Solomon G, Niv-Spector L, Gonen-Berger D, Callebaut I, Djiane J, Gertler A. Preparation of leptin antagonists by site-directed mutagenesis of human, ovine, rat, and mouse leptin's site I.I.I.: Implications on blocking undesired leptin action in vivo. Ann NY Acad Sci 1091: 531–539, 2006 [DOI] [PubMed] [Google Scholar]
- 43. Wang T, Hartzell DL, Rose BS, Flatt WP, Hulsey MG, Menon NK, Makula RA, Baile CA. Metabolic responses to intracerebroventricular leptin and restricted feeding. Physiol Behav 65: 839–848, 1999 [DOI] [PubMed] [Google Scholar]
- 44. Widdowson PS, Upton R, Buckingham R, Arch J, Williams G. Inhibition of food response to intracerebroventricular injection of leptin is attenuated in rats with diet-induced obesity. Diabetes 46: 1782–1785, 1997 [DOI] [PubMed] [Google Scholar]
- 45. Zhang Y, Matheny M, Tumer N, Scarpace PJ. Aged-obese rats exhibit robust responses to a melanocortin agonist and antagonist despite leptin resistance. Neurobiol Aging 25: 1349–1360, 2004 [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 372: 425–432, 1994 [DOI] [PubMed] [Google Scholar]