

Abstract
Fibrosis is a pathological scarring process that leads to destruction of organ architecture and impairment of organ function. Chronic loss of organ function in most organs, including bone marrow, heart, intestine, kidney, liver, lung, and skin, is associated with fibrosis, contributing to an estimated one third of natural deaths worldwide. Effective therapies to prevent or to even reverse existing fibrotic lesions are not yet available in any organ. There is hope that an understanding of common fibrosis pathways will lead to development of antifibrotic therapies that are effective in all of these tissues in the future. Here we review common and organ-specific pathways of tissue fibrosis.
Keywords: epigenetics, fibroblasts, fibrosis, TGF-β
fibrosis is a nonphysiological scarring process, which is associated with excessive deposition of extracellular matrix (ECM) and which leads to impairment of organ function. Progression of chronic diseases in parenchymal organs such as liver, kidney, heart, and lung is associated with fibrosis, and the fibrotic process plays a principal role in the organs' detriment. Fibrosis also plays an important role in the skin (from hypertrophic scars to sclerodermia), inflammatory bowel disease (causing strictures), in joints (rheumatoid arthritis but also joint replacement), and bone marrow (myelodysplastic syndromes). Additionally, the stroma of solid tumors can be considered fibrotic tissue (25, 26, 55). In short, fibrosis plays a role in the progression of many chronic diseases. Specific therapies to halt, or even to reverse, existing tissue fibrosis are not yet available in any organ. Because of the histomorphological similarities shared by fibrosis in all organs, the concept of common tissue fibrosis pathways that could be potential therapeutic targets in all organs is an attractive one (Fig. 1). There is ample hope that mechanistic understanding of the common fibrosis pathways will lead to development of drugs that are effective in all organs in the future (122). Here we summarize the common pathways as well as the organ-specific aspects of tissue fibrosis, which will be discussed in greater detail in specialized articles in this Theme series.
Fig. 1.
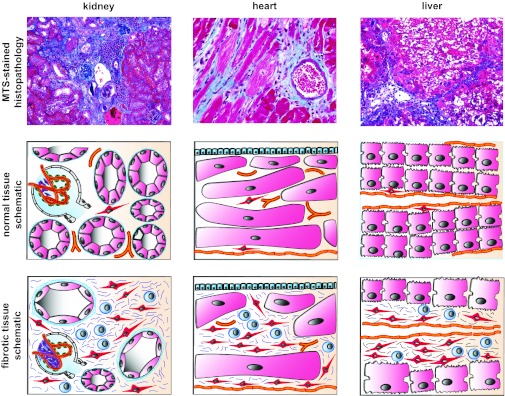
Common features of tissue fibrosis. Macroscopically and microscopically fibrotic organs share obvious commonalities, which has led to the concept of common fibrotic pathways: fibrotic organs are stiff [reflecting excessive extracellular matrix (ECM) deposition], are pale (reflecting rarefication of the vasculature), and have an uneven surface (reflecting fibroblast contraction). Histopathological analysis reveals that tissue fibrosis is unequivocally associated with injury of the parenchyme, accumulation of fibrillar ECM, accumulation of fibroblasts, rarefication of the microvasculature, and a mononuclear infiltrate. The figure illustrates the common appearance of fibrotic kidney, heart, and liver. The photomicrographs display representative MTS-stained sections of fibrotic mouse kidney, heart, and lung. Schematics illustrate the common mechanisms of tissue fibrosis: each organ consists of a functional parenchyme (pink, tubular epithelium in kidney, cardiomyocytes in heart, and hepatocytes in liver) and a connective tissue compartment containing microvessels (orange) and fibroblasts (red, and stellate cells in the liver). Fibrosis is unequivocally associated with expansion of the connective tissue compartment. The fibrotic connective tissue contains ECM fibers (blue), activated fibroblasts (red), and a mononuclear infiltrate (blue). The question is whether common or organ-specific mechanisms lead to this uniform appearance.
Activated Fibroblasts
Excessive deposition of ECM fibers is the eponymous lesion of fibrosis, and since Virchow's work in the 19th century it is known that fibroblasts are the principal source of ECM in fibrosis (115). For this reason, fibroblasts are a focus of attention in fibrosis research and the inhibition of fibroblast-mediated ECM synthesis is a principal goal of antifibrotic therapeutic approaches. Nevertheless, fibroblasts have remained relatively elusive in molecular terms and, as a consequence, therapies to specifically target fibroblasts in fibrosis are not yet available (55). While fibroblasts are easy to culture as spindle-shaped cells and are widely utilized in biomedical sciences, their identification in vivo is challenging (55). Traditionally, fibroblasts in vivo were identified in the light microscope based on their location (within connective tissue) and their spindle-shaped cytoplasm and in electron microscopy based on their leaflet-like cytoplasm, prominent endoplasmatic reticulum, and prominent cytoskeleton (55, 131). In fibrosis, the principal source of collagen is “activated fibroblasts” (reflecting their increased biosynthetic and proliferative activities). Phenotypically, activated fibroblasts are characterized by a pronounced rough endoplasmatic reticulum, stress fibers, and a large nucleolus. Since the observation that the majority of collagen-producing fibroblasts are labeled with antibodies to the filament α-smooth muscle actin (αSMA; 55), the terms “activated fibroblasts” and “myofibroblasts” are used synonymously to refer to the fibroblasts that mediate fibrosis. However, fibroblasts are a heterogenous, specialized cell type and it has become evident that a more distinctive view on specialized fibroblast populations, requiring specific markers, is required (55).
Fibroblasts not only differ among organs, they also display heterogenous phenotypes within single organs (i.e., in the kidney, lipid-laden fibroblasts from medulla differ from those in the cortex; 37), they fulfill specialized functions [a subpopulation of kidney fibroblasts is the principal source of erythropoietin (74, 88), and select atrial fibroblasts are constituents of the cardiac conduction system (16, 113)], and dermal fibroblasts can be traced to their original position from the body axis based on their Hox gene code (17). Molecular analysis of fibroblasts is particularly challenging, because a common marker that is specific to all fibroblasts does not exist (55). Markers that are commonly used to identify fibroblasts in vivo [i.e., αSMA, vimentin, fibroblast-specific protein 1 (FSP1), Desmin] only detect subpopulations of fibroblasts and none of them is specific for fibroblasts (for review see Refs. 55 and 108). Distinct contributions of these fibroblast populations to fibrosis are not yet known (55). In this regard, it is entirely unknown whether fibroblasts switch expression of their marker profile of whether detected fibroblasts are truly distinct populations. While myofibroblasts (particularly αSMA-expressing) are considered principal mediators of fibrogenesis (in the liver, ∼60% of myofibroblasts actively express collagen; 42, 75), the only fibroblast population that has been mechanistically proven to contribute to fibrogenesis is FSP1+ fibroblasts (ablation of FSP1+ fibroblasts in FSP1-tk transgenic mice ameliorates kidney fibrosis; 53). The field is further complicated by the fact that fibroblasts display different phenotypes and proliferative capacities with age, and it has been proposed that fibroblasts can differentiate along phenotypic stages similar to myleoid cells (9). Overall, analysis of fibroblasts is hindered by the lack of sufficient markers (particularly surface markers), and the evolving fibroblast heterogeneity has possibly contributed to the failure to develop fibroblast-specific therapeutic targets.
While there is a consensus that (activated) fibroblasts are the principal source of collagen and prominent mediators of fibrogenesis, it is not yet clear where these activated fibroblasts originate from (the fibroblast morphology does not offer known clues about their origin). In the traditional view, activated fibroblasts in healing wounds and fibrotic lesions derive from the resident fibroblasts through activation and proliferation. Exclusive contribution of this was first challenged by the observation that bone marrow-derived fibrocytes are recruited and contribute to fibrogenesis (15). Furthermore, epithelial cells are considered to contribute to fibroblast accumulation through epithelial-mesenchymal transition (107), vascular smooth muscle cells and pericytes have been reported to shed off vessels to become connective tissue fibroblasts (72, 100), and endothelial cells contribute to fibroblast accumulation through endothelial-mesenchymal transition (EndMT; 128). Overall, assessment of the contributions of these mechanisms differs widely among studies (which utilized different fibrosis models, fate mapping strategies, and marker analysis). While it is likely that all mechanisms contribute to fibroblast accumulation under experimental conditions, their contribution to fibroblast accumulation (percentage-wise) and fibrogenesis may differ substantially among organs, and disease models and further studies will be required (12, 14, 50, 54, 59, 62, 70, 87, 92, 97, 101, 127, 136, 140; Fig. 2). It is difficult to translate findings that were generated in transgenic reporter mice to human pathologies, because, in the absence of reporter genes, fate mapping studies are not yet feasible. For example, in the kidney, there is a strong correlation of expression of epithelial-mesenchymal transition (EMT) markers within epithelial cells and progression of fibrogenesis (39, 43), but the ultimate fate of the cells cannot be assessed.
Fig. 2.

Origins of activated fibroblasts. Fibrosis is associated with accumulation of collagen-producing (activated) fibroblasts (center, multicolored). Activated fibroblasts can derive via activation and subsequent proliferation from resident fibroblasts (blue). Bone marrow-derived fibrocytes are recruited to fibrotic lesions and contribute to accumulation of collagen-producing fibroblasts (red, top left). Endothelial cells can undergo an endothelial-to-mesenchymal transition to contribute to fibroblast accumulation (green, top right). α-Smooth muscle actin (αSMA)-positive vascular smooth muscle cells and pericytes have been proposed to shed off existing vessels and to contribute to accumulation of myofibroblasts (pink, bottom right). Epithelial cells can undergo an epithelial-to-mesenchymal transition and contribute to fibroblast accumulation (yellow, bottom left).
Microvasculature and Hypoxia
Fibrotic tissues in general appear macroscopically pale, reflecting microvascular rarefication and diminished perfusion. As a result of decreased microvasculature, fibrotic tissues are often chronically hypoxic, which directly contributes to fibrogenesis through HIF1α-mediated signaling (23, 44, 77). Mechanistically, fibrogenesis is characterized by an impaired capacity to regenerate (or form new) vessels. Fibrosis is often associated with decreased VEGF levels, and upregulation of endogenous molecules with antiangiogenic activity and VEGF administration has been shown to be beneficial in experimental models of organ fibrosis (18, 51, 73, 125). How the endothelial cells are lost in the first place, however, is not clear yet, as low incidence of endothelial-cell apoptosis does not add up with the severity of microvascular rarification (118, 125). EndMT is an additional mechanism that contributes to depletion of microvessels (94, 127–129). EndMT was originally observed in cardiac development, where mesenchymal cells (which form the atrioventricular cushion, the primordia of the valves and septa of the adult heart) are derived from the endocardium by an EndMT (27, 71, 83). We demonstrated that in the context of fibrosis of the adult heart, endothelial cells acquire a mesenchymal phenotype [in response to transforming growth factor-β1 (TGF-β)], contributing to rarefication of the microvasculature and fibroblast accumulation (128). Inhibition of EndMT ameliorated experimental fibrosis in heart and kidney (70, 128, 129). In summary, microvascular injury is an important common constituent of organ fibrosis and a promising therapeutic target.
Sterile inflammation.
Fibrosis in most cases is associated with inflammation. While the accompanying infiltrate can be reflective of a specific immune response to an underlying infection (i.e., fibrosis due to viral hepatitis or schistosomiasis in the liver or due to bacterial pyelonephritis in kidney), the prototypical fibrosis is associated with a sterile inflammation, likely in response to occurring cell death (5, 104). If inflammation is a facultative constituent of fibrogenesis or if the fibroproliferative response and inflammatory response are separate entities is a matter of debate and reviewed elsewhere (82, 121). Of note, antiinflammatory regimens are remarkably ineffective to halt fibrogenesis in clinical application, arguing that the fibroproliferative response is (at least in advanced stages) an independent process from the inflammatory reaction.
Parenchymal Injury
Per definition a scar substitutes for lost parenchymal cells, and fibrosis is invariably associated with progressive loss of the affected organ's parenchyma (i.e., hepatocytes in the liver, nephrons in kidney, cardiomyocytes in heart). While parenchymal cells possess the capacity to regenerate per se, such regenerative ability is lost during fibrosis, triggering formation of the fibrotic scar (3, 6, 102, 124). In this regard, cross talk of the injured epithelium with fibroblasts and inflammatory cells is of foremost relevance for triggering the fibrotic process. In addition to release of profibrotic metabolites (i.e., reactive oxygen species), injured parenchymal cells govern fibrogenesis through secretion of chemokines and growth factors. The prototypical profibrotic growth factor, which plays a role in fibrogenesis in all organs, is TGF-β.
Transforming Growth Factor-β
TGF-β was originally identified as a factor that induces the capacity of anchorage-independent growth, a hallmark of malignant transformation, in embryonic kidney fibroblasts (95). The prominent role of TGF-β in fibrogenesis was first observed when subcutaneous injection of purified TGF-β induced fibrotic lesions at the injection site and was further corroborated by the observation that neutralization of TGF-β with antiserum ameliorated experimental fibrosis in the kidney, and in heart and liver as well (11, 20, 64, 96). TGF-β is overexpressed in all fibrotic tissues, and it induces collagen production in cultured fibroblasts irrespective of their origin (1, 45, 60, 99). To our knowledge, the pivotal relevance of TGF-β for tissue fibrosis has not yet been disputed in any organ, making TGF-β a principal target for potential antifibrotic therapies (66). However, fibrosis (i.e., in the liver) can occur independent of TGF-β, for example mediated through an IL-13-mediated pathway (56).
Genetics and epigenetics.
During the past three decades considerable progress was made in identifying pathways that orchestrate tissue fibrosis. This knowledge, however, still offers little insight to explain the vastly different progression rates of fibrosis in individual patients with common underlying diseases and comorbidities. The root of the problem, i.e., why profibrotic pathways are more active in one patient than another, can be conceptually explained by their distinct genetic and epigenetic backgrounds (120). As it is well established that the genetic background of mice and rats (or other model organisms) affects the progression of fibrosis, it is highly likely that genetic polymorphisms equally impact fibrogenesis in human patients. However, it is proving to be difficult to link genetic polymorphisms to distinct disease progression rates. Numerous studies have been undertaken to explore the link between TGF-β—the prototypical fibrotic growth factor—and fibrosis susceptibility in a biased manner. Overall, several studies demonstrated association between TGF-β polymorphisms in kidney, lung, liver, and skin keloids, whereas multiple other studies failed to document such association (40, 57, 58, 76, 86, 91, 93, 110, 123). Most studies focused on single-nucleotide polymorphism (SNP) rs1982073, a T-to-C transition of the 29th nucleotide in the coding sequence, which results in a leucine-to-proline substitution at codon 10 (L10P) and which has been associated with elevated TGF-β1 levels (139). This polymorphism has been found to be associated with increased predisposition to develop renal fibrosis in patients with diabetes mellitus (89), with increased risk to develop chronic cardiac graft failure (46), and with increased risk to develop liver cirrhosis in patients with hepatitis C infection (117). However, other studies did not reveal such association between TGF-β1 SNPs and fibrosis risk (69, 126), possibly a reflection of the generally minor impact of SNPs on activity of their gene products. Owing to the failure to link fibrosis progression rates to single SNPs involving obvious genes (with established mechanistic impact of fibrogenesis), currently, numerous efforts are underway to identify patterns of multiple SNPs that correlate with fibrosis progression in genome-wide association studies (GWAS).
Another attractive explanation for distinct risks for developing fibrosis is epigenetics. The best studied fibrosis-associated phenomenon with regard to epigenetics is the maintenance of an activated phenotype when fibroblasts are explanted from fibrotic tissues (while fibroblast activation associated with physiological repair is reversible; 2, 80, 98, 131). Aberrant promoter methylation of specific genes plays a role in maintaining the activated phenotype (10). We identified Rasal1, which encodes for a suppressor of the Ras oncoprotein to be selectively hypermethylated in fibrotic kidney fibroblasts (10). Since Rasal1 is also hypermethylated in activated hepatic stellate cells, the speculation that Rasal1 epigenetic silencing is part of a common fibrotic program is an attractive concept (112). In addition to impacting fibrotic fibroblast activation, epigenetics plays a role in the progressive loss of the parenchymal regenerative capacity (i.e., through histone modifications in tubular epithelial cells which cause cell-cycle arrest) or modulation of inflammatory cells. While epigenetic modifications in general are acquired in response to environmental stimuli, evidence is increasing that epigenetics also plays a role in determining the inherited susceptibility to develop fibrosis. For example, multigenerational transmittance of histone modifications at TGF-β1 and PPARγ loci can lower the susceptibility to develop liver fibrosis (138).
Organ-Specific Aspects of Fibrosis
While fibrosis follows similar pathways across different organs, justifying the term “tissue fibrosis,” each organ has unique features of fibrosis as well. Unique, organ-specific features of fibrosis are discussed from our personal experience below (Table 1).
Table 1.
Fibrogenesis in kidney, liver, heart, and lung
| Kidney | Liver | Heart | Lung | |
|---|---|---|---|---|
| Spontaneous repair of acute injury | ++ | +++ | (+) | (+) |
| Spontaneous repair of fibrotic lesions | (+) | ++ | (+) | (+) |
| Association of fibrosis with infection | (+) | ++ | (+) | + |
| Fibrosis leading to cancer | (+) | ++ | + |
Albeit similar appearance of fibrotic lesions, tissue fate differs substantially among organs. Liver displays by far the best regenerative capacity. Liver cirrhosis is considered a precancerous stage, and an association between pulmonary fibrosis and cancer exists but not in kidney and heart. Liver fibrosis is mostly due to viral infection; in kidney, heart, and lung, infections are not a common cause of fibrosis.
Skin fibrosis.
Fibrosis of the skin is the integral component of a variety of heterogenous disorders including hypertrophic scars, keloids, and scleroderma (85). Hypertrophic scar formation is the best established (earliest known documentation of excessive scarring dates back to 1700 BC in the Smith papyrus), most common (an estimated 50 million patients suffer from excessive scar formation following surgery or trauma annually in developed countries) prototypical fibrotic lesion (35). As opposed to physiological scars, hypertrophic scars are raised above skin level (but are confined to the original wound) and they are often associated with pain and itching and cause disfigurement (35). In contrast, keloids are thick scar tissues, which “have escaped the boundaries of the original wound” (35). Keloids occur in all races, but black Africans are particularly susceptible. Unlike these trauma-associated localized fibrotic lesions, skin fibrosis (then referred as scleroderma) is the leading manifestation of systemic sclerosis, a rare connective tissue disease, which manifests with widespread fibrosis of the skin, but also lungs and other internal organs such as heart and kidney and widespread vasculopathy (38). Furthermore, scleroderma can manifest locally (then referred to as localized scleroderma), often in context with drug or toxin exposure, radiation, or metabolic disorders. The scleroderma, which is associated with systemic sclerosis, is particularly intriguing, as it is the leading manifestation of a systemic fibrotic syndrome, possibly highlighting the common pathways of tissue fibrosis (67, 78, 114). For example, analysis of systemic sclerosis reiterated the common impact of TGF-β1 on fibrogenesis.
Liver fibrosis.
The liver stands out from all other tissues with regard to its capacity to regenerate fibrotic lesions, and based on our experience with animal models it is not surprising that reports of reversibility of fibrotic lesions in humans originated from observations of liver patients (Table 2). Because of the high prevalence of hepatitis virus infections, liver cirrhosis may well be the clinically most relevant form of tissue fibrosis worldwide. However, the strong association of infections and liver fibrosis suggests that other pathways (as opposed to the mostly noninfectious causes in other organs) may be at play in the liver. Furthermore, the liver has a unique capacity to regenerate per se as is known since the myth of Prometheus. As a result of the unique dynamics of liver fibrosis, use of Picro Sirius Red Staining or transmission electron microscopy to identify more cross-linked (and thus more stable) collagen fibers is especially useful for fibrosis assessment in the liver. The liver is also unique in the regard that stellate cells (neuro-crest-derived, lipid-laden cells) are a major source of myofibroblasts (through a process often referred to as transdifferentiation) as opposed to the “activation” of “common” fibroblasts (which are also present as “periportal fibroblasts”) in the liver.
Table 2.
Specific aspects of organ fibrosis
| Kidney | Liver | Heart | Lung | Skin | |
|---|---|---|---|---|---|
| Top 3 etiologies | Diabetes mellitus | Viral hepatitis | Hypertension | Idiopathic pulmonary fibrosis (IPF) | Physical injury |
| Hypertension | Alcohol-induced | Coronary artery disease | Occupational diseases | Idiopathic scleroderma | |
| Glomerulonephritis | Nonalcoholic-steatohepatitis (NASH) | Aortic stenosis | Sarcoidosis | ||
| Epidemiology | Prevalence of chronic kidney disease ≈5% of general population (US) | Prevalence of liver cirrhosis: ≈ 5–10% of general population | Prevalence of diastolic heart failure ≈ 1% of the general population | Prevalence 0.03% of general population | Prevalence of hypertrophic scars ≤75% upon physical trauma |
| Prevalence of end-stage renal disease ≈0.2% of general population | Prevalence of systemic scleroderma ≈ 0.02% of general population | ||||
| Diagnosis | Blood tests to assess excretory kidney function (serum creatinine, BUN, GFR) | Blood tests to assess global liver function | Functional assessment of diastolic dysfunction through Doppler echocardiography or invasive measurements (i.e., LVEDP ) | Imaging (HRCT scan, chest X-ray) | Location (association with prior lesion) |
| Kidney biopsy | Imaging (FibroScan, ARFI, MRI) | Spirometry (restriction and impaired gas exchange) | Appearance (i.e., pigmentation, aesthetics) | ||
| Liver biopsy | Lung biopsy | Stiffness, elasticity skin thickness (SScl) | |||
| Relevant classifications of fibrosis | Area of kidney cortex occupied by fibrotic tissue (rel. %), glomeruli viewed as separate entity (glomerulosclerosis) | Distinction between fibrosis (early) and cirrhosis | Distinction between endomyocardial and perivascular fibrosis | Radiographic location and appearance (central vs. peripheral; upper lobes vs. lower lobes) | Adequate scar vs. hypertrophic scar vs. keloid |
| Grading for inflammatory activity | Local vs. systemic scleroderma | ||||
| Clinical reports of regression of established fibrosis | Regression of fibrosis in patients with diabetic nephropathy 10 yr after pancreas transplantation (31, 32) | Liver fibrosis regresses upon removal of the pathogen (21, 30, 49, 65, 105, 106) | Sporadic reports of improved diastolic dysfunction and regression of cardiac fibrosis in patients with hypertensive heart disease upon blood pressure normalization (13) | Case reports of pulmonary fibrosis regression upon treatment or removal of underlying pathogens | Hypertrophic scars regress regularly, only sporadic reports of keloid regression |
| Sporadic reports of improved kidney function upon intensified ACE inhibitor therapy (103) | Reports of regression of cirrhosis are debated (28, 52, 90) | ||||
| Unique features | Kidney fibrosis causes anemia due to cessation of erythropoietin production when renal fibroblasts become activated | Stellate cells contribute to liver fibrosis | Because cardiac fibroblasts are required for electromechanic coupling, fibrosis can cause atrial fibrillation | Mean time of survival of patients with IPF is <6 yr | Association of skin color and keloid formation (none in albinos, highest incidence in African population) |
| Microvascular shunts are detrimental, because they cause porto-venous hypertension |
ACE, angiotensin-converting enzyme; ARFI, acoustic radiation force impulse imaging; BUN, blood urea nitrogen; GFR, glomerular filtration rate; HRCT, high-resolution computed tomography; LVEDP, left ventricular end diastolic pressure.
Cardiac fibrosis.
The heart stands out with regard to its distinct types of fibrotic lesions. Anatomically, “perivascular fibrosis” can be distinguished from interstitial (“endomyocardial”) and subendocardial fibrosis. So-called endocardial fibroelastosis (EFE) is a unique form of fibrosis found in newborns with congenital heart defects, which is characterized by subendocardial deposition of elastic fibers (29, 34, 63). Unlike in other organs discussed here, the cardiac parenchymal cells are muscle cells (cardiomyocytes) and not epithelial cells, displaying a very limited regenerative capacity. As a consequence of the absent regenerative ability of cardiomyocytes, extensive scarring (“physiological fibrosis”) is necessary to seal the heart following myocardial infarction, and the distinction between “physiological fibrosis” and “pathological fibrosis” is more evident in the heart than in any other tissue (19, 130). Cardiac fibroblasts are unique, as they also function as mechanoelectrical transducers (16, 113). Hence, the physiological role of fibroblasts (the most abundant cell type in the heart) is more obvious than in any other organ discussed here. Among all organs, assessment of fibrosis in live animals (or patients for that matter) is most challenging, because only functional measurement of diastolic dysfunction (disturbed relaxation of the heart due to increased stiffness) correlates with fibrosis, whereas biochemical blood tests that correspond with heart fibrosis are not available. As a result, cardiac fibrosis is still less appreciated in the clinical setting compared with fibrosis in liver and kidney, for example.
Kidney fibrosis.
In the kidney we found the disparity between the renal capacity to regenerate severe acute injury and the irreversibility of fibrotic lesions to be most striking: while denuded tubular basement membranes can be repopulated without scar formation, kidney fibrosis without treatment is really irreversible. One reason may be the complexity of kidney architecture. With regard to kidney fibrosis, glomeruli and tubulointerstitium are viewed as separate entities: fibrosis of glomeruli (referred to as “glomerulosclerosis”) typically precedes the classic fibrosis involving the connective tissue between the tubules. Compared with the heart, fibroblasts in the normal kidney are relatively scarce. A subset of kidney fibroblasts is the principal source of erythropoietin in the body, possibly explaining the link between fibrosis and anemia in chronic kidney disease (erythropoietin expression is lost when fibroblasts become activated myofibroblasts; 7, 74, 88).
Lung fibrosis.
Fibrosis of the lung causes primarily restrictive ventilator defects. While the fibrosis that is associated with most chronic lung pathologies (i.e., obstructive pulmonary diseases, emphysema) is not the primary determinant of a patient's prognosis, generalized progressive pulmonary fibrosis is particularly devastating (84, 119). The two most intriguing forms are idiopathic pulmonary fibrosis and the pulmonary fibrosis as part of the systemic sclerosis (scleroderma) manifestation, leading to death of affected patients within two to six years (81). Compared with fibrotic lesions in other organs, relatively mild fibrotic lesions are fatal. While both idiopathic pulmonary fibrosis and systemic sclerosis-associated interstitial lung disease (SSc-ILD) are considered to follow similar pathways, antiinflammatory treatment is the standard for SSc-ILD, whereas antiinflammatory therapy rather worsens the prognosis of idiopathic pulmonary fibrosis (81, 121).
Implications of Antifibrotic Therapies for Common Pathways of Fibrogenesis
While the concept of common druggable organ fibrosis pathways is highly attractive, it is proving difficult to single out such relevant common pathways. As discussed above, “organ fibrosis” is not a monocausal disease, but rather an umbrella for a collection of chronic pathologies in multiple organs that are dependent on diverse and complex interactions of various cell types. Owing to the complexity and variability of organ fibrosis, it is difficult to establish a hierarchy according to relevance of involved mechanisms, possibly contributing to the failure to establish antifibrotic therapies for clinical application as yet. Nevertheless, numerous strategies have been proven to be successful in multiple models of organ fibrosis and there is additional information that can be gathered from failed clinical trials, to determine which pathway is truly relevant, and which is not. Among the most promising strategies to inhibit progression of fibrogenesis are modulation of the accompanying inflammation, pharmacological prevention of ECM deposition, correction of the altered epigenome, and the most obvious—inhibition of profibrotic growth factors (for review see Ref. 122). In this regard, inhibition of TGF-β1 ameliorated fibrogenesis in fibrosis models involving skin, liver, kidney, lung, and heart and clinical trials with neutralizing antibodies are underway involving systemic sclerosis, myelofibrosis and diabetic nephropathy (122). However, direct inhibition of TGF-β1 is not without risk, as it possesses strong antiinflammatory activity (TGF-β1-knockout mice display generalized inflammation) and mutations of genes encoding for constituents of the TGF-β1 signaling pathway can be directly linked to gastrointestinal cancers.
In our experience, analysis of the antifibrotic potential of bone morphogenic protein-7 (BMP7), a morphogen with TGF-β1-neutralizing activity, provided valuable insights into the common, functionally relevant pathways of organ fibrosis. BMP7 (synonym osteogenic protein-1, OP1) is the prototypical member of the family of bone morphogenic proteins, which, as members of the TGF-β superfamily, signal through activation of activin-like kinase receptors and Smad transcription factors (135, 137). Our own interest in BMP7 stems from screening experiments in which we identified BMP7 as a potent inhibitor of TGF-β1-induced epithelial-mesenchymal transition (EMT). In subsequent studies, our group and others found administration of recombinant BMP7 to inhibit fibrosis in rodent fibrosis involving kidney (24, 47, 79, 109, 116, 132, 133), liver (61, 136), heart (48, 128), and colon (33), allowing for identification of mechanisms that are relevant for fibrogenesis across all organs. BMP7 inhibits ECM production by fibroblasts (132), decreases chemokine release by injured kidney epithelial cells (36), and inhibits EMT and EndMT programs (128, 133), reaffirming that these TGF-β1 signaling-controlled events are relevant common fibrosis pathways.
Future Directions - Reversal of Fibrosis
Deemed unthinkable a decade ago, several preclinical and clinical studies suggested that fibrosis cannot only be stopped, but that regeneration of fibrotic organs is possible. The most obvious evidence for regression of fibrotic lesions is the remodeling that occurs during maturation of hypertrophic scars. The strongest evidence for reversal of fibrosis involving parenchymal internal organs was provided in the liver, where several clinical studies reported resolution of fibrosis upon successful treatment of underlying hepatitis infections (30, 49). Regression of kidney fibrosis was reported in patients with underlying diabetic nephropathy upon successful pancreas transplantation (31, 32). Numerous studies reported possible regression of established fibrosis in animal models of fibrogenesis. Common to all reports of reversed fibrosis is that supplementation of matrix-degrading compounds was not necessary, suggesting that fibrotic ECM is turned over and that endogenous mechanisms for removal of deposited ECM exist (i.e., through macrophages). Furthermore, it is becoming evident that regeneration of the chronically injured parenchyme needs to be a cornerstone of potential fibrosis-reversing strategies. Currently, there are three principal strategies emerging to regenerate lost parenchyma: the best established strategies are cell-based approaches in which pluri- or mulipotent progenitor cells are provided to repair injured organs (4, 8, 22, 68, 111). Other strategies are to stimulate the endogenous regenerative capacity. In this regard, supplementation of morphogens such as BMPs or Wnt proteins, which are believed to recreate the embryonic microenvironment, have been proven to be promising in preclinical studies (41, 133, 134). A third strategy involves correction of the aberrant epigenetics of chronically injured cells, to possibly rescue their physiological regenerative capacity (10, 124).
In summary, organ fibrosis is a rapidly evolving field and there is reason for optimism, that vast improvements that were made in deciphering underlying molecular mechanisms will finally translate to effective antifibrotic therapies. Tissue-specific aspects of fibrogenesis, which are needed for understanding the common features of organ fibrosis, are discussed in further detail in this THEME series.
GRANTS
R. Kalluri is supported by US National Institutes of Health (NIH) Grants DK55001, DK081576, CA125550, CA155370, CA151925, and CA163191, the Metastasis Research Center at the MD Anderson Cancer Center, and the Cancer Prevention and Research Institute of Texas. M. Zeisberg is supported by Deutsche Forschungsgemeinschaft (DFG) Grant ZE523/2-1 and the Else-Kröner Memorial Stipend 2005/59.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.Z. prepared the figures; M.Z. and R.K. drafted the manuscript; M.Z. and R.K. edited and revised the manuscript; M.Z. and R.K. approved the final version of the manuscript.
REFERENCES
- 1. Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y, Leask A. Tumor necrosis factor alpha suppresses the induction of connective tissue growth factor by transforming growth factor-beta in normal and scleroderma fibroblasts. J Biol Chem 275: 15220–15225, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol 26: 587–595, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Akram KM, Lomas NJ, Spiteri MA, Forsyth NR. Clara cells inhibit alveolar epithelial wound repair via TRAIL-dependent apoptosis. Eur Respir J. Epub ahead of print doi:10.1183/09031936.00213411 [DOI] [PubMed] [Google Scholar]
- 4. Akram KM, Samad S, Spiteri M, Forsyth NR. Mesenchymal stem cell therapy and lung diseases. Adv Biochem Eng Biotechnol. Epub ahead of print doi:10.1007/10_2012_140 [DOI] [PubMed] [Google Scholar]
- 5. Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol 22: 1007–1018, 2011 [DOI] [PubMed] [Google Scholar]
- 6. Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, Allison M, Coleman N, Alexander G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol. Epub ahead of print doi:10.1016/j.jhep.2012.10.031 [DOI] [PubMed] [Google Scholar]
- 7. Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, Yamamura K, Nagoshi N, Shibata S, Rao TN, Fehling HJ, Fukatsu A, Minegishi N, Kita T, Kimura T, Okano H, Yamamoto M, Yanagita M. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 121: 3981–3990, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Assmus B, Fischer-Rasokat U, Honold J, Seeger FH, Fichtlscherer S, Tonn T, Seifried E, Schachinger V, Dimmeler S, Zeiher AM. Transcoronary transplantation of functionally competent BMCs is associated with a decrease in natriuretic peptide serum levels and improved survival of patients with chronic postinfarction heart failure: results of the TOPCARE-CHD Registry. Circ Res 100: 1234–1241, 2007 [DOI] [PubMed] [Google Scholar]
- 9. Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, Francz PI. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc Natl Acad Sci USA 85: 5112–5116, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med 16: 544–550, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 346: 371–374, 1990 [DOI] [PubMed] [Google Scholar]
- 12. Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J 25: 5603–5613, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102: 1388–1393, 2000 [DOI] [PubMed] [Google Scholar]
- 14. Broekema M, Harmsen MC, van Luyn MJ, Koerts JA, Petersen AH, van Kooten TG, van Goor H, Navis G, Popa ER. Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol 18: 165–175, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1: 71–81, 1994 [PMC free article] [PubMed] [Google Scholar]
- 16. Camelliti P, Green CR, Kohl P. Structural and functional coupling of cardiac myocytes and fibroblasts. Adv Cardiol 42: 132–149, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA 99: 12877–12882, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, Housset C, Rosmorduc O. Hypoxia-induced VEGF, collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 35: 1010–1021, 2002 [DOI] [PubMed] [Google Scholar]
- 19. Czubryt MP. Common threads in cardiac fibrosis, infarct scar formation, and wound healing. Fibrogenesis Tissue Repair 5: 19, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Denis M. Neutralization of transforming growth factor-beta 1 in a mouse model of immune-induced lung fibrosis. Immunology 82: 584–590, 1994 [PMC free article] [PubMed] [Google Scholar]
- 21. Dienstag JL, Goldin RD, Heathcote EJ, Hann HW, Woessner M, Stephenson SL, Gardner S, Gray DF, Schiff ER. Histological outcome during long-term lamivudine therapy. Gastroenterology 124: 105–117, 2003 [DOI] [PubMed] [Google Scholar]
- 22. Dimmeler S, Zeiher AM. Wanted! The best cell for cardiac regeneration. J Am Coll Cardiol 44: 464–466, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, Michel BA, Gay RE, Muller-Ladner U, Matucci-Cerinic M, Plate KH, Gassmann M, Gay S. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res 95: 109–116, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Dube PH, Almanzar MM, Frazier KS, Jones WK, Charette MF, Paredes A. Osteogenic Protein-1: gene expression and treatment in rat remnant kidney model. Toxicol Pathol 32: 384–392, 2004 [DOI] [PubMed] [Google Scholar]
- 25. Dvorak HF, Form DM, Manseau EJ, Smith BD. Pathogenesis of desmoplasia. I. Immunofluorescence identification and localization of some structural proteins of line 1 and line 10 guinea pig tumors and of healing wounds. J Natl Cancer Inst 73: 1195–1205, 1984 [PubMed] [Google Scholar]
- 26. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315: 1650–1659, 1986 [DOI] [PubMed] [Google Scholar]
- 27. Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res 77: 1–6, 1995 [DOI] [PubMed] [Google Scholar]
- 28. Fallowfield JA, Kendall TJ, Iredale JP. Reversal of fibrosis: no longer a pipe dream? Clin Liver Dis 10: 481–497, 2006 [DOI] [PubMed] [Google Scholar]
- 29. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis, and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5: 15, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, Scioscia R, Serra G, Lai ME, Loy M, Caruso L, Desmet V, Purcell RH, Balestrieri A. Long-term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology 126: 1740–1749, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 339: 69–75, 1998 [DOI] [PubMed] [Google Scholar]
- 32. Fioretto P, Sutherland DE, Najafian B, Mauer M. Remodeling of renal interstitial and tubular lesions in pancreas transplant recipients. Kidney Int 69: 907–912, 2006 [DOI] [PubMed] [Google Scholar]
- 33. Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem 285: 20202–20212, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Friehs I, Illigens B, Melnychenko I, Zhong-Hu T, Zeisberg E, Del Nido PJ. An animal model of endocardial fibroelastosis. J Surg Res. Epub ahead of print doi:10.1016/j.jss.2012.07.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med 17: 113–125, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gould SE, Day M, Jones SS, Dorai H. BMP7 regulates chemokine, cytokine, and hemodynamic gene expression in proximal tubule cells. Kidney Int 61: 51–60, 2002 [DOI] [PubMed] [Google Scholar]
- 37. Grupp C, Lottermoser J, Cohen DI, Begher M, Franz HE, Muller GA. Transformation of rat inner medullary fibroblasts to myofibroblasts in vitro. Kidney Int 52: 1279–1290, 1997 [DOI] [PubMed] [Google Scholar]
- 38. Hawk A, English JC., 3rd Localized and systemic scleroderma. Semin Cutan Med Surg 20: 27–37, 2001 [DOI] [PubMed] [Google Scholar]
- 39. Hazzan M, Hertig A, Buob D, Copin MC, Noel C, Rondeau E, Dubois-Xu YC. Epithelial-to-mesenchymal transition predicts cyclosporine nephrotoxicity in renal transplant recipients. J Am Soc Nephrol 22: 1375–1381, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Helmig S, Belwe A, Schneider J. Association of transforming growth factor beta1 gene polymorphisms and asbestos-induced fibrosis and tumors. J Investig Med 57: 655–661, 2009 [DOI] [PubMed] [Google Scholar]
- 41. Hendry CE, Little MH. Reprogramming the kidney: a novel approach for regeneration. Kidney Int 82: 138–146, 2012 [DOI] [PubMed] [Google Scholar]
- 42. Herbst H, Frey A, Heinrichs O, Milani S, Bechstein WO, Neuhaus P, Schuppan D. Heterogeneity of liver cells expressing procollagen types I and IV in vivo. Histochem Cell Biol 107: 399–409, 1997 [DOI] [PubMed] [Google Scholar]
- 43. Hertig A, Bonnard G, Ulinski T, Colombat M, Jouanneau C, Baugey E, Bensman A, Ronco P, Rondeau E, Xu-Dubois YC. Tubular nuclear accumulation of Snail and epithelial phenotypic changes in human myeloma cast nephropathy. Hum Pathol 42: 1142–1148, 2011 [DOI] [PubMed] [Google Scholar]
- 44. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117: 3810–3820, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hogaboam CM, Blease K, Mehrad B, Steinhauser ML, Standiford TJ, Kunkel SL, Lukacs NW. Chronic airway hyperreactivity, goblet cell hyperplasia, and peribronchial fibrosis during allergic airway disease induced by Aspergillus fumigatus. Am J Pathol 156: 723–732, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holweg CT, Baan CC, Balk AH, Niesters HG, Maat AP, Mulder PM, Weimar W. The transforming growth factor-beta1 codon 10 gene polymorphism and accelerated graft vascular disease after clinical heart transplantation. Transplantation 71: 1463–1467, 2001 [DOI] [PubMed] [Google Scholar]
- 47. Hruska KA, Guo G, Wozniak M, Martin D, Miller S, Liapis H, Loveday K, Klahr S, Sampath TK, Morrissey J. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol 279: F130–F143, 2000 [DOI] [PubMed] [Google Scholar]
- 48. Hua JY, Zhang ZC, Jiang XH, He YZ, Chen P. [Relationship between endothelial-to-mesenchymal transition and cardiac fibrosis in acute viral myocarditis]. Zhejiang Da Xue Xue Bao Yi Xue Ban 41: 298–304, 2012 [PubMed] [Google Scholar]
- 49. Hui CK, Leung N, Shek TW, Yao H, Lee WK, Lai JY, Lai ST, Wong WM, Lai LS, Poon RT, Lo CM, Fan ST, Lau GK. Sustained disease remission after spontaneous HBeAg seroconversion is associated with reduction in fibrosis progression in chronic hepatitis B Chinese patients. Hepatology 46: 690–698, 2007 [DOI] [PubMed] [Google Scholar]
- 50. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte andnot epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ioannou M, Pyrpasopoulou A, Simos G, Paraskeva E, Nikolaidou C, Venizelos I, Koukoulis G, Aslanidis S, Douma S. Upregulation of VEGF expression is associated with accumulation of HIF-1alpha in the skin of naive scleroderma patients. Mod Rheumatol. Epub ahead of print doi:10.1007/s10165-012-0787-6 [DOI] [PubMed] [Google Scholar]
- 52. Ismail MH, Pinzani M. Reversal of liver fibrosis. Saudi J Gastroenterol 15: 72–79, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM, Neilson EG. Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts. Mol Ther 3: 149–159, 2001 [DOI] [PubMed] [Google Scholar]
- 54. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341–350, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 6: 392–401, 2006 [DOI] [PubMed] [Google Scholar]
- 56. Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, Wakefield LM, Letterio JJ, Wynn TA. IL13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol 173: 4020–4029, 2004 [DOI] [PubMed] [Google Scholar]
- 57. Khalil MS, El Nahas AM, Blakemore AI. Transforming growth factor-beta1 SNPs: genetic and phenotypic correlations in progressive kidney insufficiency. Nephron Exp Nephrol 101: e31–e41, 2005 [DOI] [PubMed] [Google Scholar]
- 58. Kikuchi K, Tanaka A, Matsushita M, Kitazawa E, Hosoya N, Kawashima Y, Selmi C, Gershwin ME, Miyakawa H. Genetic polymorphisms of transforming growth factor beta-1 promoter and primary biliary cirrhosis in Japanese patients. Ann NY Acad Sci 1110: 15–22, 2007 [DOI] [PubMed] [Google Scholar]
- 59. Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, Chapman HA. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest 119: 213–224, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim Y, Ratziu V, Choi SG, Lalazar A, Theiss G, Dang Q, Kim SJ, Friedman SL. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem 273: 33750–33758, 1998 [DOI] [PubMed] [Google Scholar]
- 61. Kinoshita K, Iimuro Y, Otogawa K, Saika S, Inagaki Y, Nakajima Y, Kawada N, Fujimoto J, Friedman SL, Ikeda K. Adenovirus-mediated expression of BMP-7 suppresses the development of liver fibrosis in rats. Gut 56: 706–714, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol 45: 429–438, 2006 [DOI] [PubMed] [Google Scholar]
- 63. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 225: 631–637, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 106: 130–135, 2002 [DOI] [PubMed] [Google Scholar]
- 65. Kweon YO, Goodman ZD, Dienstag JL, Schiff ER, Brown NA, Burchardt E, Schoonhoven R, Brenner DA, Fried MW. Decreasing fibrogenesis: an immunohistochemical study of paired liver biopsies following lamivudine therapy for chronic hepatitis B. J Hepatol 35: 749–755, 2001 [DOI] [PubMed] [Google Scholar]
- 66. Leask A, Abraham DJ. TGFbeta signaling and the fibrotic response. FASEB J 18: 816–827, 2004 [DOI] [PubMed] [Google Scholar]
- 67. Leask A, Denton CP, Abraham DJ. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol 122: 1–6, 2004 [DOI] [PubMed] [Google Scholar]
- 68. LeBleu V, Sugimoto H, Mundel TM, Gerami-Naini B, Finan E, Miller CA, Gattone VH, 2nd, Lu L, Shield CF, 3rd, Folkman J, Kalluri R. Stem cell therapies benefit Alport syndrome. J Am Soc Nephrol 20: 2359–2370, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lee JJ, Park SK, Kwon OS, Won IS, Kim DK, Jung YK, Ku YS, Kim YS, Choi DJ, Kim JH. Genetic polymorphism at codon 10 of the transforming growth factor-beta1 gene in patients with alcoholic liver cirrhosis. Korean J Hepatol 17: 37–43, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59: 2612–2624, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 166: 359–367, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maeshima Y, Makino H. Angiogenesis and chronic kidney disease. Fibrogenesis Tissue Repair 3: 13, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maxwell PH, Osmond MK, Pugh CW, Heryet A, Nicholls LG, Tan CC, Doe BG, Ferguson DJ, Johnson MH, Ratcliffe PJ. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 44: 1149–1162, 1993 [DOI] [PubMed] [Google Scholar]
- 75. Milani S, Herbst H, Schuppan D, Kim KY, Riecken EO, Stein H. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology 98: 175–184, 1990 [DOI] [PubMed] [Google Scholar]
- 76. Mittal RD, Manchanda PK. Is low-frequency distribution of TGF-beta genotype associated with increased risk for end-stage renal disease? DNA Cell Biol 26: 172–177, 2007 [DOI] [PubMed] [Google Scholar]
- 77. Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol 296: G582–G592, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Morgan-Rowe L, Nikitorowicz J, Shiwen X, Leask A, Tsui J, Abraham D, Stratton R. Thrombospondin 1 in hypoxia-conditioned media blocks the growth of human microvascular endothelial cells and is increased in systemic sclerosis tissues. Fibrogenesis Tissue Repair 4: 13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J Am Soc Nephrol 13, Suppl 1: S14–S21, 2002 [PubMed] [Google Scholar]
- 80. Muller GA, Rodemann HP. Characterization of human renal fibroblasts in health and disease: I. Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proven interstitial fibrosis. Am J Kidney Dis 17: 680–683, 1991 [DOI] [PubMed] [Google Scholar]
- 81. Murray LA, Rubinowitz A, Herzog EL. Interstitial lung disease: is interstitial lung disease the same as scleroderma lung disease? Curr Opin Rheumatol 24: 656–662, 2012 [DOI] [PubMed] [Google Scholar]
- 82. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat Rec 258: 119–127, 2000 [DOI] [PubMed] [Google Scholar]
- 84. Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122: 2756–2762, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. O'Leary R, Wood EJ, Guillou PJ. Pathological scarring: strategic interventions. Eur J Surg 168: 523–534, 2002 [PubMed] [Google Scholar]
- 86. Oliver J, Agundez JA, Morales S, Fernandez-Arquero M, Fernandez-Gutierrez B, de la Concha EG, Diaz-Rubio M, Martin J, Ladero JM. Polymorphisms in the transforming growth factor-beta gene (TGF-beta) and the risk of advanced alcoholic liver disease. Liver Int 25: 935–939, 2005 [DOI] [PubMed] [Google Scholar]
- 87. Osterreicher CH, Penz-Osterreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, Sasik R, Hardiman G, Karin M, Brenner DA. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci USA 108: 308–313, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Paliege A, Rosenberger C, Bondke A, Sciesielski L, Shina A, Heyman SN, Flippin LA, Arend M, Klaus SJ, Bachmann S. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int 77: 312–318, 2010 [DOI] [PubMed] [Google Scholar]
- 89. Patel A, Scott WR, Lympany PA, Rippin JD, Gill GV, Barnett AH, Bain SC: The TGFbeta 1 gene codon 10 polymorphism contributes to the genetic predisposition to nephropathy in Type 1 diabetes. Diabet Med 22: 69–73, 2005 [DOI] [PubMed] [Google Scholar]
- 90. Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology 50: 1294–1306, 2009 [DOI] [PubMed] [Google Scholar]
- 91. Prasad P, Tiwari AK, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK. Association of TGFbeta1, TNFalpha, CCR2 and CCR5 gene polymorphisms in type-2 diabetes and renal insufficiency among Asian Indians. BMC Med Genet 8: 20, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int 80: 41–50, 2011 [DOI] [PubMed] [Google Scholar]
- 93. Radwan MI, Pasha HF, Mohamed RH, Hussien HI, El-Khshab MN. Influence of transforming growth factor-beta1 and tumor necrosis factor-alpha genes polymorphisms on the development of cirrhosis and hepatocellular carcinoma in chronic hepatitis C. patients. Cytokine 60: 271–276, 2012 [DOI] [PubMed] [Google Scholar]
- 94. Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi C. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol 179: 2660–2673, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Roberts AB, Anzano MA, Lamb LC, Smith JM, Frolik CA, Marquardt H, Todaro GJ, Sporn MB. Isolation from murine sarcoma cells of novel transforming growth factors potentiated by EGF. Nature 295: 417–419, 1982 [DOI] [PubMed] [Google Scholar]
- 96. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Faucit AS. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA 83: 4167–4171, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 108: E1475–E1483, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rodemann HP, Muller GA. Characterization of human renal fibroblasts in health and disease: II. In vitro growth, differentiation, and collagen synthesis of fibroblasts from kidneys with interstitial fibrosis. Am J Kidney Dis 17: 684–686, 1991 [DOI] [PubMed] [Google Scholar]
- 99. Romanelli RG, Caligiuri A, Carloni V, DeFranco R, Montalto P, Ceni E, Casini A, Gentilini P, Pinzani M. Effect of pentoxifylline on the degradation of procollagen type I produced by human hepatic stellate cells in response to transforming growth factor-beta 1. Br J Pharmacol 122: 1047–1054, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest 95: 859–873, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, Weiss SJ. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol Cell Biol 31: 2392–2403, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 287: 1253–1258, 2000 [DOI] [PubMed] [Google Scholar]
- 103. Ruggenenti P, Perticucci E, Cravedi P, Gambara V, Costantini M, Sharma SK, Perna A, Remuzzi G. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 19: 1213–1224, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257, 2007 [DOI] [PubMed] [Google Scholar]
- 105. Serpaggi J, Carnot F, Nalpas B, Canioni D, Guechot J, Lebray P, Vallet-Pichard A, Fontaine H, Bedossa P, Pol S. Direct and indirect evidence for the reversibility of cirrhosis. Hum Pathol 37: 1519–1526, 2006 [DOI] [PubMed] [Google Scholar]
- 106. Shiratori Y, Imazeki F, Moriyama M, Yano M, Arakawa Y, Yokosuka O, Kuroki T, Nishiguchi S, Sata M, Yamada G, Fujiyama S, Yoshida H, Omata M. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Ann Intern Med 132: 517–524, 2000 [DOI] [PubMed] [Google Scholar]
- 107. Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130: 393–405, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther 5: 1640–1646, 2006 [DOI] [PubMed] [Google Scholar]
- 109. Sugimoto H, LeBleu VS, Bosukonda D, Keck P, Taduri G, Bechtel W, Okada H, Carlson W, Jr, Bey P, Rusckowski M, Tampe B, Tampe D, Kanasaki K, Zeisberg M, Kalluri R. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med 18: 396–404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Suzuki S, Tanaka Y, Orito E, Sugauchi F, Hasegawa I, Sakurai M, Fujiwara K, Ohno T, Ueda R, Mizokami M. Transforming growth factor-beta-1 genetic polymorphism in Japanese patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol 18: 1139–1143, 2003 [DOI] [PubMed] [Google Scholar]
- 111. Takami T, Terai S, Sakaida I. Advanced therapies using autologous bone marrow cells for chronic liver disease. Discov Med 14: 7–12, 2012 [PubMed] [Google Scholar]
- 112. Tao H, Huang C, Yang JJ, Ma TT, Bian EB, Zhang L, Lv XW, Jin Y, Li J. MeCP2 controls the expression of RASAL1 in the hepatic fibrosis in rats. Toxicology 290: 327–333, 2011 [DOI] [PubMed] [Google Scholar]
- 113. Tveito A, Lines G, Artebrant R, Skavhaug O, Maleckar MM. Existence of excitation waves for a collection of cardiomyocytes electrically coupled to fibroblasts. Math Biosci 230: 79–86, 2011 [DOI] [PubMed] [Google Scholar]
- 114. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117: 557–567, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Virchow R. Die Cellularpathologie in ihrer Begruendung auf physiologische und pathologische Gewebelehre. Edited by Hirschwald A. Berlin, Germany: 1858 [Google Scholar]
- 116. Vukicevic S, Basic V, Rogic D, Basic N, Shih MS, Shepard A, Jin D, Dattatreyamurty B, Jones W, Dorai H, Ryan S, Griffiths D, Maliakal J, Jelic M, Pastorcic M, Stavljenic A, Sampath TK. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J Clin Invest 102: 202–214, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wang H, Mengsteab S, Tag CG, Gao CF, Hellerbrand C, Lammert F, Gressner AM, Weiskirchen R. Transforming growth factor-beta1 gene polymorphisms are associated with progression of liver fibrosis in Caucasians with chronic hepatitis C infection. World J Gastroenterol 11: 1929–1936, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Worda M, Sgonc R, Dietrich H, Niederegger H, Sundick RS, Gershwin ME, Wick G. In vivo analysis of the apoptosis-inducing effect of anti-endothelial cell antibodies in systemic sclerosis by the chorionallantoic membrane assay. Arthritis Rheum 48: 2605–2614, 2003 [DOI] [PubMed] [Google Scholar]
- 119. Wright JL, Tazelaar HD, Churg A. Fibrosis with emphysema. Histopathology 58: 517–524, 2011 [DOI] [PubMed] [Google Scholar]
- 120. Wynn TA. Fibrosis under arrest. Nat Med 16: 523–525, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 208: 1339–1350, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Xaubet A, Marin-Arguedas A, Lario S, Ancochea J, Morell F, Ruiz-Manzano J, Rodriguez-Becerra E, Rodriguez-Arias JM, Inigo P, Sanz S, Campistol JM, Mullol J, Picado C. Transforming growth factor-beta1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 168: 431–435, 2003 [DOI] [PubMed] [Google Scholar]
- 124. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, Hanley A, Isner JM, Losordo DW. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 111: 2073–2085, 2005 [DOI] [PubMed] [Google Scholar]
- 126. Yu SK, Kwon OS, Jung HS, Bae KS, Kwon KA, Kim YK, Kim YS, Kim JH. Influence of transforming growth factor-beta1 gene polymorphism at codon 10 on the development of cirrhosis in chronic hepatitis B virus carriers. J Korean Med Sci 25: 564–569, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67: 10123–10128, 2007 [DOI] [PubMed] [Google Scholar]
- 128. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13: 952–961, 2007 [DOI] [PubMed] [Google Scholar]
- 129. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res 107: 1304–1312, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zeisberg M, Strutz F, Muller GA. Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol 13, Suppl 3: S111–S120, 2000 [PubMed] [Google Scholar]
- 132. Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Muller GA, Kalluri R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol 285: F1060–F1067, 2003 [DOI] [PubMed] [Google Scholar]
- 133. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP7 counteracts TGFbeta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9: 964–968, 2003 [DOI] [PubMed] [Google Scholar]
- 134. Zeisberg M, Kalluri R. Experimental strategies to reverse chronic renal disease. Blood Purif 22: 440–445, 2004 [DOI] [PubMed] [Google Scholar]
- 135. Zeisberg M. Bone morphogenic protein-7 and the kidney: current concepts and open questions. Nephrol Dial Transplant 21: 568–573, 2006 [DOI] [PubMed] [Google Scholar]
- 136. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282: 23337–23347, 2007 [DOI] [PubMed] [Google Scholar]
- 137. Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci 13: 6991–6998, 2008 [DOI] [PubMed] [Google Scholar]
- 138. Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, Oakley F, Burt AD, Wilson CL, Anstee QM, Barter MJ, Masson S, Elsharkawy AM, Mann DA, Mann J. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med 18: 1369–1377, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zheng W. Genetic polymorphisms in the transforming growth factor-beta signaling pathways and breast cancer risk and survival. Methods Mol Biol 472: 265–277, 2009 [DOI] [PubMed] [Google Scholar]
- 140. Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol 338: 251–261, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]