

Abstract
Sorcin localizes in cellular membranes and has been demonstrated to modulate cytosolic Ca2+ handling in cardiac myocytes. Sorcin also localizes in mitochondria; however, the effect of sorcin on mitochondrial Ca2+ handling is unknown. Using mitochondrial pericam, we measured mitochondrial Ca2+ concentration and fluxes in intact neonatal cardiac myocytes overexpressing sorcin. Our results showed that sorcin increases basal and caffeine-stimulated mitochondrial Ca2+ concentration. This effect was associated with faster Ca2+ uptake and release. The effect of sorcin was specific for mitochondria, since similar results were obtained with digitonin-permeabilized cells, where cytosolic Ca2+ flux was disrupted. Furthermore, mitochondria of cardiac myocytes in which sorcin was overexpressed were more Ca2+-tolerant. Experiments analyzing apoptotic signaling demonstrated that sorcin prevented 2-deoxyglucose-induced cytochrome c release. Furthermore, sorcin prevented hyperglycemia-induced cytochrome c release and caspase activation. In contrast, antisense sorcin induced caspase-3 activation. Thus, sorcin antiapoptotic properties may be due to modulation of mitochondrial Ca2+ handling in cardiac myocytes.
Keywords: pericam, gene transfer, calcium oscillations, adenovirus, sorcin overexpression, apoptosis
sorcin, a 22-kDa Ca2+-binding protein member of the penta EF-hand family (19), was initially identified in multidrug-resistant cells (20, 23, 36). Subsequently, sorcin has been detected in a wide variety of mammalian tissues, including heart and skeletal muscle (22). Sorcin induces a drug-resistant phenotype and shows antiapoptotic properties in human colorectal cancer cells, although the mechanism is incompletely understood (17, 18).
Sorcin translocates from the cytosol to membranes upon binding of Ca2+. Translocation takes place at micromolar Ca2+ concentrations and is reversed when the cation concentration is lowered by addition of EGTA (24, 37). Translocation from the cytosol to membranes allows sorcin to interact with specific target proteins. Sorcin is primarily localized to endoplasmic reticulum and mitochondria in neuronal cells (29). In cardiac cells, sorcin localizes to junctions between the transverse tubule system and junctional sarcoplasmic reticulum (SR) and to mitochondria only at points directly in contact with or in close proximity to the SR near transverse tubules (22). Furthermore, we found that expression of sorcin was increased in the mitochondrial fraction from adult cardiac myocytes overexpressing sorcin (35). However, the function of sorcin in mitochondria is unknown. Recently, evidence that sorcin has an important role in modulating cardiac contractility through its effects on ryanodine receptor (RyR2), L-type Ca2+ channels, or Na+/Ca2+ exchanger (NCX) has accumulated (21, 33, 35). We demonstrated that cardiac overexpression of sorcin, achieved via adenoviral gene therapy techniques, reverses cardiac dysfunction in diabetic mice by improving cytosolic Ca2+ handling (35). Taken together, these findings suggest a role for sorcin as a Ca2+-handling-modulating protein.
Although interaction of sorcin with plasma membrane and SR proteins has been studied, the role of sorcin in mitochondria is completely unexplored. Participation of mitochondria in regulation of cytosolic Ca2+ may be minimal. However, cytosolic Ca2+ seems to have a major role in modulating mitochondrial energy production by stimulating dehydrogenases and ATP synthase (12). Furthermore, excess Ca2+ accumulation in mitochondria is a common event in the process of cell death by necrosis and apoptosis (8, 31). Therefore, changes in mitochondrial Ca2+ concentration ([Ca2+]m) are an important signal in cellular physiology. However, measurement of [Ca2+]m in the intact cell has been difficult because of the practical problem of distinguishing signals originating from mitochondria from those originating from cytoplasmic locales. Despite these difficulties, beat-to-beat oscillations of [Ca2+]m have been reported using Ca2+ indicators that are not specific for mitochondria. Recently, the ratiometric Ca2+-sensitive yellow fluorescent protein pericam, selectively expressed in mitochondria, was used in a study showing beat-to-beat Ca2+ oscillations in neonatal cardiac myocytes (32). Using targeted mitochondrial pericam after sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) overexpression, we successfully determined mitochondrial Ca2+ (2).
The goal of this work was to determine whether sorcin modulates [Ca2+]m homeostasis in intact neonatal rat cardiac myocytes and explore whether the effects of sorcin on [Ca2+]m are part of the mechanisms of sorcin-induced antiapoptotic effects. Using adenoviral gene transfer of sorcin, we measured [Ca2+]m with ratiometric pericam. Our results show, for the first time, that sorcin increases [Ca2+]m by activating Ca2+ uptake and increasing mitochondrial Ca2+ tolerance, preventing Ca2+-induced cytochrome c release and hyperglycemia-induced apoptosis.
MATERIALS AND METHODS
All animal protocols were approved by the University of California, San Diego, Institutional Animal Care and Use Committee and conform to the Guide for the Care and Use of Laboratory Animals as outlined by the National Institutes of Health.
Cardiac myocyte culture and adenoviral infection.
Rat neonatal cardiac myocytes were isolated as described previously (13). Briefly, ventricles from 1- to 2-day-old neonatal rats were minced, digested with collagenase and pancreatin, and subjected to discontinuous Percoll (Pharmacia LKB Biotechnology) gradient centrifugation. The myocyte-enriched fraction was washed twice and resuspended in culture medium. Cells were plated onto gelatin-coated culture dishes or glass chamber slides. Culture medium consisted of 4:1 DMEM-M199, 10% horse serum, 5% fetal bovine serum, and 1% penicillin-streptomycin-amphotericin B (Fungizone). Final glucose concentration was 20 mM. After 24 h of plating, myocytes were infected with a multiplicity of infection of 20 for all viruses. Studies were performed after 48 h of viral infection.
Cardiac myocytes exposed to high glucose.
Cells were allowed to adhere to the plates for 24 h before change to basic experimental culture medium (4.5:1 DMEM-M199, 2% fetal bovine serum, and 1% penicillin-streptomycin-amphotericin B, supplemented with glucose) at physiological [normal (5.5 mM)] or elevated [high (25 mM)] glucose concentrations and maintained in these conditions for 72 h. Cells were subjected to viral infection 24 h after high-glucose exposure.
Construction of adenoviral vectors.
Generation of sorcin adenovirus (Adv-sorcin) has been described previously (35). We cloned a mouse gene coding for sorcin (GenBank accession no. BF178692) into a replication-deficient adenoviral vector under control of the promoter-enhancer region of the human cytomegalovirus (Adv-sorcin). An empty adenovirus without transgene (Adv-control) was used in the control group. Antisense sorcin (AS-sorcin) was generated by cloning the sorcin cDNA in reverse orientation relative to the promoter, as described previously (14).
Mitochondrial pericam has been constructed and described by Nagai et al. (28). Pericam is a circularly permuted yellow fluorescent family protein that was fused to calmodulin (CaM) and M13, a 26-residue peptide derived from the CaM-binding region of the skeletal muscle myosin. The Ca2+-bound CaM and M13 peptide form a stable and compact complex. We used the ratiometric pericam, which has a bimodal excitation spectrum peaking at 415 and 494 nm. Pericam was targeted specifically to the mitochondrial matrix (mitochondrial pericam) by fusion to a cytochrome oxidase sequence.
We cloned the gene coding for mitochondrial pericam into a replication-deficient adenoviral vector under control of the promoter-enhancer region of the human cytomegalovirus (Adv-CMVmpericam). Cells were infected with Adv-CMVmpericam using 20 plaque-forming units per cell.
Pericam fluorescence.
Cells cultured on a chambered cover glass were perfused with HEPES-buffered medium containing 1.8 mM CaCl2 at room temperature. Chambers were mounted in a Nikon Diaphot epifluorescence microscope equipped with a ×100 Fluor objective (oil immersion) interfaced to a dual-excitation lamp system (Solamere Technologies, Salt Lake City, UT) set at 410 nm for excitation 1 and 485 nm for excitation 2 via a set of filters. Fluorescence emission was filtered at 510 nm and directed to a photomultiplier tube. Additionally, an aperture mechanism allowed fluorescence to be collected from a selected portion of the field, which was always positioned over the peripheral, nonnuclear regions of individual cells. Data were collected from the emission channel at a rate of 20 Hz, and the ratio of intensity at 410-nm excitation to intensity at 485-nm excitation was calculated providing for relative comparisons of the [Ca2+]m between experimental treatments. [Ca2+]m was determined as follows: [Ca2+]m = Kd (R − Rmin)/(Rmax − R), where R represents the fluorescence intensity ratio Fλ1/Fλ2 [λ1 (∼410 nm) and λ2 (∼485 nm) are the fluorescence detection wavelengths for pericam]. Ratios corresponding to the titration end points are denoted by subscripts indicating the minimum (min) and maximum (max) Ca2+ concentration. Kd is the Ca2+ dissociation constant of ratiometric pericam, which is 1.7 μM (28). Rmax was measured in situ by application of 5 mM Ca2+ in the presence of ionophore (ionomycin). Rmin was obtained by application of EGTA (9).
Indo 1 fluorescence.
The indo 1-facilitated Ca2+ transient after adenoviral infection was measured as described previously (35).
Preparation of subcellular fractions.
Mitochondria and SR microsomes were prepared using differential centrifugation, as described previously (35). Myocytes were homogenized with a Polytron in a buffer containing 30 mM Tris, 300 mM sucrose, and protease inhibitor cocktail (1:1,000 dilution; Sigma). The first centrifugation was performed at 1,500 g for 15 min at 4°C, and the pellet containing the nuclear fraction and cellular debris was discarded. The supernatant was spun at 8,000 g for 15 min. The pellet containing mitochondria was washed twice and spun twice at the same speed. The supernatant was spun for 1 h at 160,000 g. The pellet was used as the SR microsome fraction. Fractions were kept frozen at −80°C until they were tested.
Western blot analysis.
Myocytes were homogenized with a Polytron in lysis buffer. Protein content was measured by the Bradford method (Bio-Rad) and adjusted for equal loading. Protein extracts (20 μg) were separated by a 4–12% Bis-Tris·HCl-buffered polyacrylamide gel (Invitrogen, Carlsbad, CA) and subjected to Western blotting. SERCA2a and cytochrome c antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The primary antibody for Western blotting of sorcin was a rabbit anti-sorcin polyclonal antibody (a kind gift from Dr. Hector Valdivia, Dept. of Physiology, University of Wisconsin Medical School, Madison, WI). Antibodies against caspase-9, caspase-3, cleaved caspase-3, poly(ADP-ribose)polymerase (PARP), and cleaved PARP were obtained from Cell Signaling Technology (Danvers, MA). The secondary antibody was a horseradish peroxidase-conjugated anti-rabbit. The blots were also probed by a mouse monoclonal porin (voltage-dependent anion channel) antibody or actin antibody as internal controls to ensure equivalent protein loading and protein integrity. Signals on the films were digitized on a 350-dots/inch scanner.
Analysis of mitochondrial Ca2+ transients.
Resting [Ca2+]m was obtained from myocytes that did not present spontaneous contraction and were not paced. Diastolic and systolic [Ca2+]m are the minimum and maximum pericam ratios, respectively, that were measured during contraction. Ca2+ uptake is the maximum slope of the upstroke. Ca2+ release is the maximum slope during Ca2+ decline. Δ[Ca2+]m is the difference between diastolic and systolic [Ca2+]m.
Statistical analysis.
Values are means ± SE from at least three different experiments. Comparisons between means were analyzed, as appropriate, by Student's t-test or one-way ANOVA followed by Bonferroni's post test. P < 0.05 was considered statistically significant.
RESULTS
[Ca2+]m in neonatal cardiac myocytes.
Results from a typical experiment in which pericam was used to measure [Ca2+]m are shown in Fig. 1A. [Ca2+]m in resting cells was 255 ± 27 nM. [Ca2+]m increased immediately after electrical stimulation of the cell at 0.3 Hz, and beat-to-beat oscillations were observed (Fig. 1A). After stimulation was stopped, [Ca2+]m slowly returned to resting levels (Fig. 1A). Interestingly, increasing the rate of pacing further increased [Ca2+]m, which is in accordance with previous reports (data not shown) (4, 26).
Fig. 1.

Expression of sorcin in mitochondria and sarcoplasmic reticulum (SR) of cardiac myocytes and its effect on mitochondrial Ca2+. A: typical trace of pericam ratio [ratio of excitation at 485 nm to excitation at 410 nm (485/410)] from a cell infected with control adenovirus (Adv-control, bottom trace) and a cell infected with sorcin adenovirus (Adv-sorcin, top trace). Cells were paced at 0.3 Hz. B: Western blot analysis of sorcin, SERCA2a, and voltage-dependent anion channel (porin) in mitochondria and SR from cells infected with Adv-control (C) or Adv-sorcin (S).
Effect of sorcin overexpression on [Ca2+]m.
Adv-sorcin transfer in neonatal cardiac myocytes resulted in a dramatic increase in sorcin protein in mitochondrial, as well as SR, fractions (Fig. 1B). This increase in mitochondrial sorcin was not due to contamination from the SR fraction, since SERCA2a was minimal in the mitochondrial fraction (Fig. 1B). [Ca2+]m was higher in cardiac myocytes infected with Adv-sorcin than in cells infected with Adv-control (Fig. 2, A and B). Sorcin increased diastolic (60%) and systolic (80%) [Ca2+]m (Fig. 2B). The magnitude of the mitochondrial Ca2+ transient was increased approximately twofold by sorcin (Fig. 2C).
Fig. 2.

Analysis of mitochondrial Ca2+ transients of sorcin-overexpressing cells. A: typical recording of Ca2+ transients from a cell infected with Adv-control and a cell infected with Adv-sorcin. B: cumulative diastolic and systolic mitochondrial Ca2+ concentrations ([Ca2+]m) in control and sorcin-overexpressing cells. C: magnitude of the Ca2+ transient (difference between diastolic and systolic Ca2+). D: effect of sorcin on mitochondrial Ca2+ uptake and release. Values are means ± SE, n = 20 cells per group. *P < 0.05 vs. control.
Effect of sorcin on mitochondrial Ca2+ fluxes.
Sorcin overexpression stimulated mitochondrial Ca2+ uptake by 150% (Fig. 2D). Sorcin also stimulated Ca2+ release by 250% (Fig. 2D). Caffeine induces SR Ca2+ release via RyR2 and is able to increase [Ca2+]m by increasing cytosolic Ca2+ in microdomains (34). Therefore, we investigated the effect of sorcin on caffeine-induced [Ca2+]m increase. Figure 3A shows a typical recording of cytosolic Ca2+ assessed with indo 1. Recordings of caffeine-induced Ca2+ release from SR in a control cell and a sorcin-infected cell are shown. Ca2+ release by the sorcin-infected cell was increased. Figure 3B shows the effect of 10 mM caffeine on [Ca2+]m in a cell infected with Adv-control and a cell infected with Adv-sorcin. Caffeine induced a slow rise in pericam signal in the control cell; however, the Adv-sorcin-infected cell presented an accelerated mitochondrial Ca2+ uptake with a higher maximum peak than the control cell (Fig. 3B).
Fig. 3.

Effect of sorcin on stimulated mitochondrial Ca2+ uptake in intact and permeabilized cardiac myocytes. A: cytosolic Ca2+ in a representative experiment in which caffeine-perfused intact myocytes were treated with Adv-control or Adv-sorcin. B: mitochondrial Ca2+ in a representative experiment in which caffeine-infused intact myocytes were treated with Adv-control or Adv-sorcin. C: effect of sorcin on Ca2+ uptake in permeabilized cardiac myocytes. Cells were incubated with 25 μM digitonin for 30 min in buffer containing 5 mM EGTA. Buffer was changed to medium containing 50 μM Ca2+. Inset: fraction of the curve's upstroke with an expanded time scale to show differences in the slope.
Effect of sorcin on [Ca2+]m of permeabilized cardiac myocytes.
To determine whether sorcin has a direct effect on the mitochondria, we permeabilized myocytes with 25 μM digitonin for 30 min and subsequently challenged mitochondria with 50 μM Ca2+. This manipulation induces a rapid Ca2+ uptake by the mitochondria, increasing [Ca2+]m, which eventually will promote opening of the transition pore and other mechanisms of Ca2+ release that lead to a decrease in [Ca2+]m, as shown at the end of the curves. Figure 3C shows a typical recording of the experiment. Ca2+ uptake was more rapid in sorcin-overexpressing than control cells (Fig. 3C, inset). The maximum Ca2+ uptake was higher in the cells that overexpressed sorcin (Fig. 3C).
Sorcin reduces cytochrome c release and caspase activation.
Sorcin overexpression in cardiac cells increased mitochondrial protein levels of cytochrome c and decreased cytosolic cytochrome c levels (Fig. 4). Cytochrome c can be detected in the cytosolic fraction of control cells, because the normal culture medium contains 20 mM glucose, which has been demonstrated to induce apoptosis (5, 16). To investigate if sorcin could reduce cytochrome c release during apoptosis-induced stimulus, we incubated cardiac myocytes with 3 mM 2-deoxyglucose (DOG) for 24 h (27). Figure 5 shows that DOG reduced mitochondrial and increased cytosolic cytochrome c protein levels, which is in agreement with results reported by others (27). Sorcin effectively reduced cytochrome c release (Fig. 5). Hyperglycemia is known to induce cytochrome c and trigger apoptosis in cardiac myocytes (5, 16). Thus we sought to investigate the effects of sorcin on glucose-induced apoptosis. Figure 6 shows cytosolic cytochrome c levels assessed by Western blotting. High glucose (25 mM) dramatically increased (by 7-fold) cytochrome c. Sorcin expression significantly decreased cytochrome c levels, despite high glucose. Caspase-9, caspase-3, and PARP are activated by high glucose, as demonstrated in Fig. 7A by Western blotting. Densitometric analysis (Fig. 7B) showed that high glucose reduced protein levels of caspase-9, caspase-3, and PARP. In contrast, cleaved caspase-3 and cleaved PARP were increased by high glucose. Sorcin expression in myocytes exposed to high glucose reduced caspase activation, as demonstrated by increased caspase-9, caspase-3, and PARP protein levels. In addition, cleaved caspase-3 and cleaved PARP protein levels were reduced in cells expressing sorcin, despite high glucose.
Fig. 4.

Influence of sorcin on cytochrome c (Cyt c) content. Western blot shows protein levels of cytochrome c in mitochondrial and cytosolic fraction in control (C or Ctr) and sorcin-overexpressing (S or Sor) cells. Values (means ± SE, n = 4 for each group) are averages of densitometry values [arbitrary units (AU)] and were normalized with porin and actin for mitochondrial and cytosolic fractions, respectively. *P < 0.05 vs. Ctr.
Fig. 5.

Sorcin prevents cytochrome c release induced by 2-deoxyglucose (DOG). Western blot shows protein levels of cytochrome c in mitochondrial and cytosolic fractions in control cells (Ctr), cells treated with DOG, and sorcin-overexpressing cells treated with DOG (Sor + DOG). Values (means ± SE, n = 4 for each group) are averages of densitometry values of each sample and were normalized with porin and actin for mitochondrial and cytosolic fractions, respectively. *P < 0.05 vs. Ctr and Sor + DOG.
Fig. 6.

Sorcin decreases cytochrome c release induced by hyperglycemia. Western blot shows protein levels of cytochrome c in the cytosolic fraction in normal glucose (NG, 5.5 mM), high glucose (HG, 25 mM), and HG + Adv-sorcin (HG + Sor). NG- and HG-treated cells received an empty adenovirus as control. Values (means ± SE, n = 4 for each group) are averages of densitometry values of each sample and were normalized with actin. *P < 0.05 vs. NG. &P < 0.5 vs. HG.
Fig. 7.
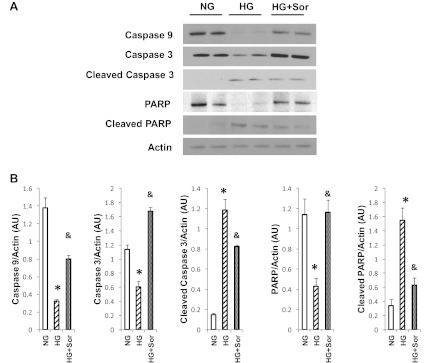
Sorcin inhibits glucose-induced caspase activation. A: Western blot showing protein levels of caspase-9, caspase-3, cleaved caspase-3, poly(ADP-ribose)polymerase (PARP), and cleaved PARP in cells exposed to normal glucose (NG, 5.5 mM glucose + 20 mM mannitol), high glucose (HG, 25 mM), and HG + Adv-sorcin (HG + Sor). B: averages of densitometry values of each sample. Values are means ± SE, n = 4 for each group. *P < 0.05 vs. NG. &P < 0.5 vs. HG.
Reducing sorcin protein levels using AS-sorcin RNA induces caspase activation.
AS-sorcin reduced sorcin protein levels by 32% (Fig. 8). In addition, caspase-3 was decreased and cleaved caspase-3 was increased, indicating activation of the caspase pathway (Fig. 8).
Fig. 8.

Antisense sorcin (AS-sorcin) promotes caspase-3 activation. Western blot shows effect of AS-sorcin on protein levels of sorcin, caspase-3, and cleaved caspase-3. Values (means ± SE, n = 4 for each group) are averages of densitometry values of each sample and were normalized with actin. *P < 0.05 vs. control.
DISCUSSION
Our findings demonstrate, for the first time, that the sorcin-antiapoptotic effect is associated with alterations in mitochondrial Ca2+ handling in neonatal cardiac myocytes. We found an increase in [Ca2+]m after adenoviral transfer of sorcin that can be due to activation of mitochondrial Ca2+ uptake by sorcin. In addition, mitochondria from cells overexpressing sorcin showed an increased tolerance to Ca2+ and decreased DOG-induced cytochrome c release, as well as inhibition of hyperglycemia-induced apoptotic signaling.
Recent work by us and others has established a role for sorcin in modulating cytosolic Ca2+ handling by affecting RyR2, NCX, SERCA2a, and L-type Ca2+ channel (1, 7, 21, 33, 35). However, despite data showing that sorcin is expressed also in mitochondria, studies on the effect of sorcin on mitochondria were not available. Accordingly, we sought to investigate the effects of sorcin on mitochondrial Ca2+ handling and whether these effects could explain the antiapoptotic effect of sorcin.
Our findings show that mitochondria are an important sorcin target, since we found a dramatic expression of sorcin in the mitochondrial fraction after Adv-sorcin infection (Fig. 1B).
Using mitochondrial pericam, we were able to measure [Ca2+]m in the intact cardiac myocyte. Furthermore, we found beat-to-beat oscillations in the neonatal cardiac myocyte, which have been observed by others (32). Mitochondrial transients are characterized by a slow Ca2+ uptake followed by slower Ca2+ release (Figs. 1A and 2A). This pattern is in accordance with that predicted by simulations by Cromptom (6) and was also observed by Robert et al. (32).
Overexpression of sorcin results in increased [Ca2+]m. This effect of sorcin can be explained by activation of Ca2+ uptake, possibly by activating the mitochondrial Ca2+ uniporter (MCU). An alternative explanation is that the higher [Ca2+]m after sorcin overexpression is reflecting an increase in cytosolic Ca2+ concentration, which may be increased by sorcin, as has been demonstrated in the adult rat cardiac myocyte (35). We observed activation of Ca2+ uptake in the intact cell and the digitonin-permeabilized myocyte. These results indicate that sorcin has a direct effect on the mitochondria. However, steady-state [Ca2+]m in sorcin-overexpressing cells may be the result of effects of sorcin on different cellular targets. Sorcin modulates cytosolic Ca2+ handling by its effects on RyR2, L-type Ca2+ channel, or NCX (21, 33, 35). A RyR isoform in mitochondria that participates in rapid Ca2+ uptake has been reported (3). In addition, it has been suggested that mitochondria can transiently experience microdomains of high Ca2+ concentration during systole (34). Furthermore, other studies have shown that the affinity of the MCU for Ca2+ is relatively low (10, 11, 15, 25) and only mitochondria in close proximity to the RyR2 of SR may be exposed to high enough Ca2+ concentrations for optimal MCU function (30). Normal Ca2+ loading of the SR and Ca2+ release by RyR2 are required for adequate mitochondrial Ca2+ uptake by the MCU. Sorcin may play a role modulating the SR-mitochondria interaction. In this work we showed that mitochondrial Ca2+ levels mirror cytosolic Ca2+ levels after caffeine-induced SR Ca2+ release and that sorcin increased cytosolic and mitochondrial Ca2+. Increased mitochondrial Ca2+ after sorcin expression can be explained, because there is more Ca2+ in Ca2+ microdomains. However, increased mitochondrial Ca2+ uptake after sorcin overexpression suggests a specific mitochondrial effect of sorcin. This effect alone could explain the increase in mitochondrial Ca2+. In fact, [Ca2+]m was higher and Ca2+ uptake was greater in permeabilized cells expressing sorcin than control cells, indicating that the effect of sorcin on mitochondrial Ca2+ uptake can lead to increased [Ca2+]m.
Sorcin was first described in multidrug-resistant cells (36). Recently, an antiapoptotic role of sorcin has been suggested (17). Our results demonstrate that mitochondria from sorcin-overexpressing cells tolerate more Ca2+ before opening of the mitochondrial transition pore. In addition, DOG-induced cytochrome c release was prevented by sorcin. We also tested the effect of sorcin on glucose-induced apoptosis. Our results demonstrate that sorcin inhibited glucose-induced cytochrome c release and caspase activation. These results support an antiapoptotic role of sorcin in cardiac myocytes. Further support was obtained in experiments using AS-sorcin to decrease sorcin protein levels. Decrease of sorcin levels in cells exposed to hyperglycemia triggered caspase-3 activation.
In conclusion, sorcin modulates mitochondrial Ca2+ handling in neonatal cardiac myocytes. Since [Ca2+]m likely influences the efficiency of mitochondrial metabolism, sorcin may help provide a link between contractile activity and cardiac energy metabolism. Furthermore, sorcin may have an antiapoptotic role in the cardiac myocyte.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-66917 and HL-52496 and American Heart Association, Western Affiliates, Grant-in-Aid 0150735. J. Suarez is a recipient of National Heart, Lung, and Blood Institute Supplement to Promote Diversity in Health-Related Research 3R01 HL-066917-10S1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.S. and W.H.D. are responsible for conception and design of the research; J.S., P.M.M., B.T.S., A.S.-R., and H.W. performed the experiments; J.S., A.S.-R., H.W., E.S.F., and W.H.D. analyzed the data; J.S. and P.M.M. interpreted the results of the experiments; J.S. prepared the figures; J.S. drafted the manuscript; J.S., B.T.S., E.S.F., and W.H.D. edited and revised the manuscript; J.S., P.M.M., B.T.S., A.S.-R., H.W., E.S.F., and W.H.D. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Miyawaki for providing the plasmid coding for the mitochondrial pericam.
REFERENCES
- 1. Ali R, Huang Y, Maher SE, Kim RW, Giordano FJ, Tellides G, Geirsson A. miR-1 mediated suppression of sorcin regulates myocardial contractility through modulation of Ca2+ signaling. J Mol Cell Cardiol 52: 1027–1037, 2012 [DOI] [PubMed] [Google Scholar]
- 2. Belke DD, Swanson E, Suarez J, Scott BT, Stenbit AE, Dillmann WH. Increased expression of SERCA in the hearts of transgenic mice results in increased oxidation of glucose. Am J Physiol Heart Circ Physiol 292: H1755–H1763, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem 276: 21482–21488, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Brandes R, Bers DM. Simultaneous measurements of mitochondrial NADH and Ca2+ during increased work in intact rat heart trabeculae. Biophys J 83: 587–604, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome c-mediated caspase-3 activation pathway. Diabetes 51: 1938–1948, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Crompton M. The role of Ca2+ in the function and dysfunction of the heart mitochondria. In: Calcium and the Heart, edited by Langer GA. New York: Raven, 1990, p. 167–199 [Google Scholar]
- 7. Farrell EF, Antaramian A, Rueda A, Gómez AM, Valdivia HH. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem 278: 34660–34666, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Giacomello M, Drago I, Pizzo P, Pozzan T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ 14: 1267–1274, 2007 [DOI] [PubMed] [Google Scholar]
- 9. Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. J Biol Chem 276: 29188–29194, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol Cell Physiol 267: C313–C339, 1994 [DOI] [PubMed] [Google Scholar]
- 11. Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol 258: C755–C786, 1990 [DOI] [PubMed] [Google Scholar]
- 12. Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem 184: 359–369, 1998 [PubMed] [Google Scholar]
- 13. Hartong R, Villarreal FJ, Giordano F, Hilal-Dandan R, McDonough PM, Dillmann WH. Phorbol myristate acetate-induced hypertrophy of neonatal rat cardiac myocytes is associated with decreased sarcoplasmic reticulum Ca2+-ATPase (SERCA2) gene expression and calcium reuptake. J Mol Cell Cardiol 28: 2467–2477, 1996 [DOI] [PubMed] [Google Scholar]
- 14. He H, Meyer M, Martin JL, McDonough PM, Ho P, Lou X, Lew WY, Hilal-Dandan R, Dillmann WH. Effects of mutant and antisense RNA of phospholamban on SR Ca2+-ATPase activity and cardiac myocyte contractility. Circulation 100: 974–980, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360–364, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Kumar S, Kain V, Sitasawad SL. High glucose-induced Ca2+ overload and oxidative stress contribute to apoptosis of cardiac cells through mitochondrial dependent and independent pathways. Biochim Biophys Acta 1820: 907–920, 2012 [DOI] [PubMed] [Google Scholar]
- 17. Landriscina M, Laudiero G, Maddalena F, Amoroso MR, Piscazzi A, Cozzolino F, Monti M, Garbi C, Fersini A, Pucci P, Esposito F. Mitochondrial chaperone Trap1 and the calcium binding protein sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res 70: 6577–6586, 2010 [DOI] [PubMed] [Google Scholar]
- 18. Maddalena F, Laudiero G, Piscazzi A, Secondo A, Scorziello A, Lombardi V, Matassa DS, Fersini A, Neri V, Esposito F, Landriscina M. Sorcin induces a drug-resistant phenotype in human colorectal cancer by modulating Ca2+ homeostasis. Cancer Res 71: 7659–7669, 2011 [DOI] [PubMed] [Google Scholar]
- 19. Maki M, Narayana SV, Hitomi K. A growing family of the Ca2+-binding proteins with five EF-hand motifs. Biochem J 328: 718–720, 1997 [PMC free article] [PubMed] [Google Scholar]
- 20. Meyers MB, Biedler JL. Increased synthesis of a low molecular weight protein in vincristine-resistant cells. Biochem Biophys Res Commun 99: 228–235, 1981 [DOI] [PubMed] [Google Scholar]
- 21. Meyers MB, Fischer A, Sun YJ, Lopes CM, Rohacs T, Nakamura TY, Zhou YY, Lee PC, Altschuld RA, McCune SA, Coetzee WA, Fishman GI. Sorcin regulates excitation-contraction coupling in the heart. J Biol Chem 278: 28865–28871, 2003 [DOI] [PubMed] [Google Scholar]
- 22. Meyers MB, Pickel VM, Sheu SS, Sharma VK, Scotto KW, Fishman GI. Association of sorcin with the cardiac ryanodine receptor. J Biol Chem 270: 26411–26418, 1995 [DOI] [PubMed] [Google Scholar]
- 23. Meyers MB, Spengler BA, Chang TD, Melera PW, Biedler JL. Gene amplification-associated cytogenetic aberrations and protein changes in vincristine-resistant Chinese hamster, mouse, and human cells. J Cell Biol 100: 588–597, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyers MB, Zamparelli C, Verzili D, Dicker AP, Blanck TJ, Chiancone E. Calcium-dependent translocation of sorcin to membranes: functional relevance in contractile tissue. FEBS Lett 357: 230–234, 1995 [DOI] [PubMed] [Google Scholar]
- 25. Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 119: 2435–2443, 2009 [DOI] [PubMed] [Google Scholar]
- 26. Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol Heart Circ Physiol 261: H1123–H1134, 1991 [DOI] [PubMed] [Google Scholar]
- 27. Most P, Boerries M, Eicher C, Schweda C, Ehlermann P, Pleger ST, Loeffler E, Koch WJ, Katus HA, Schoenenberger CA, Remppis A. Extracellular S100A1 protein inhibits apoptosis in ventricular cardiomyocytes via activation of the extracellular signal-regulated protein kinase 1/2 (ERK1/2). J Biol Chem 278: 48404–48412, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci USA 98: 3197–3202, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pickel VM, Clarke CL, Meyers MB. Ultrastructural localization of sorcin, a 22 kDa calcium binding protein, in the rat caudate-putamen nucleus: association with ryanodine receptors and intracellular calcium release. J Comp Neurol 386: 625–634, 1997 [DOI] [PubMed] [Google Scholar]
- 30. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262: 744–747, 1993 [DOI] [PubMed] [Google Scholar]
- 31. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408, 2006 [DOI] [PubMed] [Google Scholar]
- 32. Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J 20: 4998–5007, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seidler T, Miller SL, Loughrey CM, Kania A, Burow A, Kettlewell S, Teucher N, Wagner S, Kögler H, Meyers MB, Hasenfuss G, Smith GL. Effects of adenovirus-mediated sorcin overexpression on excitation-contraction coupling in isolated rabbit cardiomyocytes. Circ Res 93: 132–139, 2003 [DOI] [PubMed] [Google Scholar]
- 34. Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr 32: 97–104, 2000 [DOI] [PubMed] [Google Scholar]
- 35. Suarez J, Belke DD, Gloss B, Dieterle T, McDonough PM, Kim YK, Brunton LL, Dillmann WH. In vivo adenoviral transfer of sorcin reverses cardiac contractile abnormalities of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 286: H68–H75, 2004 [DOI] [PubMed] [Google Scholar]
- 36. Van der Bliek AM, Meyers MB, Biedler JL, Hes E, Borst P. A 22-kd protein (sorcin/V19) encoded by an amplified gene in multidrug-resistant cells is homologous to the calcium-binding light chain of calpain. EMBO J 5: 3201–3208, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zamparelli C, Ilari A, Verzili D, Vecchini P, Chiancone E. Calcium- and pH-linked oligomerization of sorcin causing translocation from cytosol to membranes. FEBS Lett 409: 1–6, 1997 [DOI] [PubMed] [Google Scholar]