

Abstract
Protegrin is an antimicrobial peptide with a β-hairpin structure stabilized by a pair of disulfide bonds. It has been extensively studied by solid-state NMR and computational methods. Here we use implicit membrane models to examine the binding of monomers on the surface and in the interior of the membrane, the energetics of dimerization, the binding to membrane pores, and the stability of different membrane barrel structures in pores. Our results challenge a number of conclusions based on previous experimental and theoretical work. The burial of monomers into the membrane interior is found to be unfavorable for any membrane thickness. Because of its imperfect amphipathicity, protegrin binds weakly, at most, on the surface of zwitterionic membranes. However, it binds more favorably onto toroidal pores. Anionic charge on the membrane facilitates the binding due to electrostatic interactions. Solid-state NMR results have suggested a parallel NCCN association of monomers in dimers and association of dimers to form octameric or decameric β-barrels. We find that this structure is not energetically plausible for binding to bilayers, because in this configuration the hydrophobic sides of two monomers point in opposite directions. In contrast, the antiparallel NCCN and especially the parallel NCNC octamers are stable and exhibit a favorable binding energy to the pore. The results of 100-ns simulations in explicit bilayers corroborate the higher stability of the parallel NCNC barrel compared with the parallel NCCN barrel. The ability to form pores in zwitterionic membranes provides a rationalization for the peptide’s cytotoxicity. The discrepancies between our results and experiment are discussed, and new experiments are proposed to resolve them and to test the validity of the models.
Introduction
Protegrins are antimicrobial peptides isolated from porcine leukocytes (1). The most studied of these is the 18-residue protegrin-1 (PG1), whose structure has been shown to be a β-hairpin stabilized by two disulfide bonds (2). It is active against Gram-positive and Gram-negative bacteria and fungi in vitro (1,3), but is also modestly hemolytic (4). Based on the fact that it forms ion channels in membranes (5,6), protegrin has been hypothesized to kill bacteria by permeabilizing their membranes. A large number of variants have been synthesized and tested for activity (7,8). One variant named IB-367 or iseganan HCl has been considered for the treatment of various conditions over the last 15 years (9–11), but a drug has not yet been approved. Synthetic mimetics of protegrins (12) and linear analogs thereof (13–16) have also been developed.
A large amount of information on the position of PG1 in membranes and its interaction with lipids has been obtained with the use of solid-state NMR (17,18). This technique has been used to determine the tilt angle in a 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) bilayer (19), the insertion depth in the membrane (20), dimerization in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (21), the topology of the dimer in POPC (22), oligomerization into a closed β-barrel in anionic lipids (23), distances between peptide Cα atoms and lipid phosphates (24), and the differential interaction of PG1 and an inactive mutant with lipopolysaccharide membranes (25). In other experimental work, oriented circular dichroism (CD) was used to detect a change in the orientation of protegrin as a function of concentration and hydration level (26), and x-ray diffraction detected protegrin-induced membrane thinning (27). Pores of protegrin were crystallized under low-hydration or low-temperature conditions, but high-resolution structure determination was not feasible (28). Studies with lipid monolayers showed that protegrin interacts much more strongly with anionic lipids (29) and phosphatidylcholine (PC) than with phosphatidylethanolamine (PE) (30).
These experimental studies have been complemented by a large number of computational studies (reviewed in Bolintineanu and Kaznessis (31)). Molecular dynamics (MD) simulations of monomers in micelles (32,33), and of monomers or dimers in bilayers, transmembrane (TM) (34–36) or on the bilayer interface (37,38), have been performed. Potentials of mean force have been calculated for dimerization (39), for tilting in the membrane interior (40), and for association and insertion into an anionic membrane (41,42). Finally, complete β-barrel models in membranes have been simulated (43–46) and ionic conductances have been estimated (47).
Despite the wealth of data available today, protegrin pore formation is not completely understood. Some of the experimental results challenge our current understanding of peptide-membrane interactions and raise further questions. For example, why would a short β-hairpin insert as a monomer in a bilayer, thin it by 10 Å, and tilt at 55° (19)? Why would the peptide form a dimer that would point different faces of the hairpin in the same direction (22), when one face is clearly more hydrophobic than the other? How does toxicity arise if protegrin inserts into PG membranes but does not insert into PC/cholesterol bilayers (23)?
In previous work using implicit membrane modeling, we found that a number of AMPs bind more strongly to pores than to flat membranes, and proposed that, at least in some cases, this may be due to their imperfect amphipathicity (48,49). Protegrin also exhibits this trait, in that one side of the hairpin has a sizeable hydrophobic cluster but is flanked by charged Arg side chains. It would be interesting, therefore, to explore the implications of this structure in terms of its function. To that end, we examined the binding of protegrin monomers, dimers, and β-barrels to membranes or membrane pores using implicit membrane models (IMMs), primarily IMM1 (50,51) but also generalized Born with a simple switching (GBSW) (52) and the Poisson-Boltzmann/surface area (PB/SA) approach. The results challenge some of the conclusions from previous experimental and theoretical work. It is hoped that they will stimulate further investigations.
Materials and Methods
In this work we used mostly IMM1 (50), which is an extension of effective energy function 1 (EEF1) for soluble proteins (53). EEF1 assumes the solvation free energy to be a sum over atomic contributions. The solvation free energy of each atom is obtained as a reference value pertaining to full solvent-exposure minus the solvation the atom loses due to the presence of surrounding atoms. The solvation free energy per unit volume is modeled as a Gaussian function. In addition, a distance-dependent dielectric (ε = r) is used for the electrostatic interactions and the ionic side chains are given a zero net charge.
IMM1 extends EEF1 to heterogeneous membrane-water systems. The membrane is considered to be parallel to the xy plane with its center at z = 0. The solvation parameters of all atoms now depend on the vertical position and are modeled as a linear combination of the values corresponding to water and to cyclohexane. The strengthening of electrostatic interactions in the membrane is accounted for by making the dielectric dependent on the vertical position. IMM1 has been extended (IMM1-GC) to include the surface potential due to the presence of anionic lipids (51), the TM voltage (54), and recently the membrane dipole potential (55).
One can model pores of different shapes (48,56) by making the pore radius a function of z; for example,
where T is the hydrophobic thickness of the membrane, Ro is the radius at the center of the pore, and k is a curvature parameter (k = 0 corresponds to a cylindrical (barrel-stave) pore, and increasing values of k give toroidal pores of increasing curvature). This model was recently extended to pores in anionic membranes (Y. He, L. Prieto, and T. Lazaridis, unpublished). Because the Gouy-Chapman equations can no longer be used, the electrostatic potential is obtained by numerical solution of the PB equation. The potential is then used as a static field in MD simulations. Explicit simulations showed that the charge density in the center of the pore drops to 60–70% of that on the flat membrane surface (Y. He, L. Prieto, and T. Lazaridis, unpublished). Thus, we adopted a drop of 60% in our calculations, with a smooth increase from the center to the edge of the pore.
We also used two other implicit membrane approaches. The first is GBSW, an IMM based on the generalized Born approximation (52) using a 30 cal/mol Å2 nonpolar surface tension coefficient. The second is the PB/SA approach, in which the electrostatic energy is obtained from the PB equation, treating the membrane as a low-dielectric continuum and the nonpolar energy is obtained by a term proportional to the solvent-exposed surface area (58,59). The PB/SA calculations were performed using the PBEQ module of CHARMM with 1003 grid points and 0.5 Å grid spacing.
The program CHARMM version c37a1 (60) was used in all calculations. The starting structures were obtained from the Protein Data Base (PDB code 1PG1 for the monomer (2) and 1ZY6 for the dimer (22)). The monomer structures were placed either at the center of the membrane with the hairpin perpendicular to the membrane (TM configuration) or at the membrane interface (center of mass at z = T/2, where T is the membrane hydrophobic thickness). For the interfacial configurations, we tried six different initial orientations, pointing toward the membrane the SS hairpin face, the hydrophobic face, the N strand, the C strand, the termini, or the turn. An energy minimization was first run with fixed backbone, followed by an energy minimization of the entire peptide, and a MD simulation at 300 K and 2 fs time step. For simulations on anionic membranes, we used an anionic fraction of 30% with area per lipid 70 Å2 and salt concentration 0.1 M. The MD simulations typically lasted 1 ns and average effective energies were computed over the last 0.6 ns. Longer simulations that were run as a check on several occasions gave very similar results. Error bars were obtained as the standard deviation of the averages computed from four sections of the production run.
Dimer simulations started from either the PDB structure of the parallel NCCN dimer or from other configurations constructed by appropriate positioning of the monomers. Nuclear Overhauser effect (NOE) constraints were used to form the desired hydrogen bonds between backbone atoms, which were then released in the production simulations. Octameric barrels were constructed in a similar way by appropriate rotations of monomers or dimers. Harmonic constraints were sometimes used on the backbone atoms to allow the side chains to relax. All constraints were released for the production simulations.
Results
Interaction of monomers with membranes
We first examine the interaction of protegrin monomers with the surface of zwitterionic and anionic lipid membranes. Looking at the structure of protegrin (Fig. 1 b), it is not immediately obvious which side would interact more favorably with the membrane. One side of the β-hairpin contains the two disulfide bonds, which are relatively apolar. The other side contains a sizeable hydrophobic cluster (Tyr, Phe, Leu, and 2 Val), which, however, is flanked by Arg residues that would hamper burial into the membrane. As discussed below, this imperfect amphipathicity of protegrin, which was recognized upon structure determination (2), may be important for its function. We clarify here that we are referring to planar amphipathicity (i.e., separation of hydrophobic and hydrophilic residues on the two sides of the hairpin) rather than radial amphipathicity (separation along the length of the hairpin). The former is relevant to interfacial binding and the latter to TM insertion. Some authors presumed that the disulfide face is the one that would interact with the membrane (37,41). In this work, we placed the NMR structure (PDB code 1PG1) at the interface of a zwitterionic or anionic membrane in six different initial orientations and ran a 1-ns MD simulation in each case.
Figure 1.
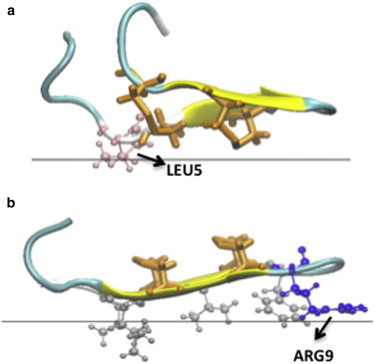
Cartoon representation in VMD (76) for the minimized final structures obtained for a neutral membrane using GBSW implicit model. The orange licorice representation indicates the position of the disulfide bonds. (a) Structure obtained when the disulfide bonds face the membrane. The pink sticks-and-balls show the position of Leu5. (b) Structure obtained when the hydrophobic cluster (silver sticks-and-balls) faces the membrane. The blue sticks-and-balls show the position of Arg9.
For the neutral membrane using IMM1, we found no binding; the peptide quickly moved away from the membrane for all six starting orientations. For the hydrophobic side, this is easily explained by repulsion of the membrane by the arginines, especially Arg-9. The disulfide side simply is not hydrophobic enough to provide sufficient stabilization. The sulfur solvation parameters in IMM1 are −1.78 kcal/mol in the membrane and −3.24 kcal/mol in water. Thus, the fact that its burial in the membrane is unfavorable is not surprising. Deprotonation of Arg-9 does not suffice to allow binding, and the peptide still moves away from the membrane.
Different results were obtained using another IMM, GBSW with the all-atom charmm22 force field. A 1-ns simulation starting from an interfacial configuration with the SS side toward the membrane did show some binding through the side chain of Leu-5 (Fig. 1 a). Although this side chain is on the other side of the hairpin, the hairpin distorts and allows partial burial of this side chain to the membrane. This provides ∼3.5 kcal/mol of membrane binding affinity. The disulfide bonds are near the interface but not buried (the z coordinates of the SG atoms are 14–16 Å, whereas the interface is at 13 Å). A simulation starting from the opposite orientation binds even more strongly, by 11.4 kcal/mol. In the optimal configuration, the hydrophobic side chains become slightly buried, whereas Arg-9 lies flat just outside the membrane core (Fig. 1 b). The difference in behavior between IMM1 and GBSW is largely due to the more-abrupt transition between nonpolar and polar regions in GBSW. This allows Arg-9 and the other arginines to maintain much of their aqueous solvation while they are close to the membrane, whereas they lose considerable solvation in IMM1.
The question of which model is closer to reality could be answered by comparison with classical biophysical experiments in solution, such as fluorescence or calorimetry. To the best of our knowledge, such experiments have not yet been performed on protegrin. However, to decide which of the two models is more correct, we could use calculations on other peptides for which such data are available. For example, it was found that buforin II does not interact at all with zwitterionic vesicles (61,62). We modeled it as an ideal helix and simulated it on a neutral membrane. Using IMM1, this peptide dissociates from the membrane in five out of six orientations, and in the simulation where it stays membrane-bound, the helix unfolds and the binding energy is −4 kcal/mol. Using GBSW, the peptide remains bound to the membrane in all simulations with an average energy of −11 kcal/mol. Thus, IMM1 seems to be in better agreement with experiment, at least for this peptide.
On the anionic membrane using IMM1-GC, binding is observed through electrostatic interactions between the positively charged residues and the negative charge of the membrane. In four out of the six simulations, the peptide points its hydrophobic side toward the membrane (Fig. 2). In the remaining two simulations, the peptide is on its side pointing the N-terminal strand toward the membrane or pointing the SS side toward the membrane. The binding energy is similar in all cases and close to 4 kcal/mol, entirely due to Arg-membrane electrostatic interactions.
Figure 2.
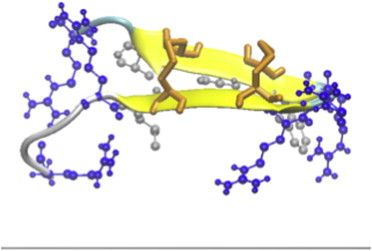
Cartoon representation in VMD for the conformation obtained most frequently in the simulations of protegrin on anionic membranes using IMM1. Orange licorice represents the disulfide bonds, and the blue and silver sticks-and-balls represent the arginines and the hydrophobic cluster, respectively.
We next considered TM configurations. A monomer was placed with its center of mass at the center of the membrane and the β-strands along the membrane normal. A 1-ns MD simulation was run to allow it to equilibrate. Table 1 shows the difference in average effective energy between the simulation in the membrane and a simulation in water as a function of membrane thickness. Transfer from water into the membrane in a TM orientation is unfavorable at all membrane thicknesses down to 10 Å. The peptide stays in the membrane during the simulation because of the high barrier of moving a number of Arg through the membrane. Similar results were obtained with the GBSW model using the same protocol, although for the thinner membranes the transfer energies were less unfavorable than those obtained with IMM1. Single-point calculations with PB/SA on energy-minimized structures obtained at the end of the GBSW simulations with ε = 2 give results between those of GBSW and IMM1 (with ε = 1 in the membrane, the transfer energies are much more unfavorable). The energy-minimized structures in all cases give less unfavorable transfer energies because the peptide can adapt its conformation to optimize its interactions. The origin of the unfavorable transfer energy in the thinnest membrane is not burial of the arginines but loss of solvation of the backbone polar groups. The contribution from the six most-buried groups that are not hydrogen bonded (6 N, 6 O, 8 N, 13 N, 13 O, and 15 N) is 14.0 kcal/mol, whereas that from the Arg side-chain N atoms is negligible. This is also evident in Fig. 3, which shows the final structure from the simulation at T = 10 Å.
Table 1.
Transfer energies (kcal/mol) from water to a TM configuration in membranes of different hydrophobic width
| T = 26 Å | T = 18 Å | T = 10 Å | |
|---|---|---|---|
| Average energies | |||
| IMM1 | +59.0 ± 2.6 | +29.5 ± 4 | +12.5 ± 2.9 |
| GBSW | +64.7 ± 1.8 | +13.4 ± 3.4 | +2.8 ± 5.2 |
| IMM1-GC | +51.2 ± 2.7 | +29.6 ± 5.1 | +5.2 ± 3.1 |
| Energy of minimized final structures | |||
| IMM1 | +44.9 | +15.5 | +0.8 |
| GBSW | +34.7 | −1.7 | −6.0 |
| PB/SA (ε = 2) | +19.4 | +7.5 | +0.3 |
| IMM1-GC | +35.7 | +16.2 | −5.9 |
Figure 3.
Final structure of a 1-ns simulation of a protegrin monomer in a 10 Å wide membrane. The blue sticks-and-balls represent the arginines. The blue and red spheres highlight the buried nonhydrogen-bonded backbone N and O atoms, respectively.
The effect of anionic membrane charge was assessed with IMM1-GC. The transfer energy is less unfavorable than in neutral membranes for T = 26 and 10 Å, and the same for T = 18 Å (Table 1). In each case, the contribution of the peptide-membrane charge interaction is ∼9 kcal/mol. The membrane charge does not reduce the transfer energy at 18 Å because Arg-9 starts to become buried at that membrane width, which costs solvation free energy. Despite the favorable contribution of the peptide-membrane charge interactions, insertion into the membrane remains unfavorable for all thicknesses, in apparent disagreement with a recent explicit PMF calculation that showed a favorable −20.1 kcal/mol free-energy change for protegrin insertion into a 1/3 POPG/PE membrane (42). These results suggest that PG1 would not get buried as a monomer in an intact membrane, even for unrealistically thin membranes.
Dimers in solution and in membranes
Protegrin is a monomer in aqueous solution (2) and apparently also in dimethyl sulfoxide (DMSO) (63). Experimental work found a tendency for the peptide to dimerize in the presence of lipids. NMR in a dodecylphosphocholine (DPC) micelle suggested an antiparallel configuration of the hairpins (64), but solid-state NMR in POPC bilayers indicated parallel NCCN dimerization (22).
First we consider dimerization in aqueous solution. There are six possible configurations: NCNC, NCCN, and CNNC, each parallel and antiparallel. All six dimers were constructed and their energy was evaluated using the EEF1 energy function. The average effective energies from a 1 ns MD simulation are given in Table 2. According to EEF1, most dimer topologies have similar energies, except for CNNC)anti. Slightly lower in energy seems to be NCCN)par. An explicit-solvent PMF calculation (39) showed NCCN)par dimerization to be more favorable than NCCN)anti by ∼12 kcal/mol, which is larger than the 6.4 kcal/mol obtained here. The GBSW model gave even larger ΔW values and structures for the dimers that are more globular than those obtained with EEF1 (the termini fold back to interact with the faces of the hairpin; results not shown). Interestingly, solid-state NMR on aggregates in aqueous buffer revealed an NCCN)par configuration (65).
Table 2.
Average effective energy (kcal/mol) per monomer in dimers of different topology
| EEF1 (water) | Neutral membrane, TM, 18 Å | ΔW(w->m) | |
|---|---|---|---|
| Monomer | −387.7 ± 2.5 | −358.2 ± 3.1 | +29.5 |
| NCCN)par | −404.8 ± 2.5 | −388.6 ± 1.0 | +16.2 |
| NCCN)anti | −401.6 ± 1.8 | −379.5 ± 2.0 | +22.1 |
| NCNC)par | −401.3 ± 2.1 | −388.8 ± 0.6 | +12.5 |
| NCNC)anti | −402.2 ± 2.2 | −384.8 ± 1.7 | +17.4 |
| CNNC)par | −400.9 ± 0.9 | −383.6 ± 1.1 | +17.3 |
| CNNC)anti | −395.9 ± 0.8 | −383.7 ± 1.4 | +12.2 |
The dimers were considered embedded within a neutral 18 Å membrane. As can be seen in Table 2, the lowest energy is observed for parallel NCCN and NCNC dimers. As expected, dimerization reduces the cost of burial into the membrane, but the transfer of all dimers from water to an 18 Å membrane is still unfavorable.
Binding of monomers to pores
We recently proposed that the imperfect amphipathicity of some AMPs may be responsible for a favorable transfer energy from flat membrane to toroidal pores, which is consistent with the idea that they stabilize the pores (48). Protegrin is an excellent illustration of this concept: a cluster of hydrophobic residues is flanked by charged residues that hamper interaction with a flat membrane, whereas a membrane surface with positive curvature allows the hydrophobic cluster to be buried while the charged residues remain solvated in water. Here we determine the energetics of binding of PG1 monomers to implicit membrane pores.
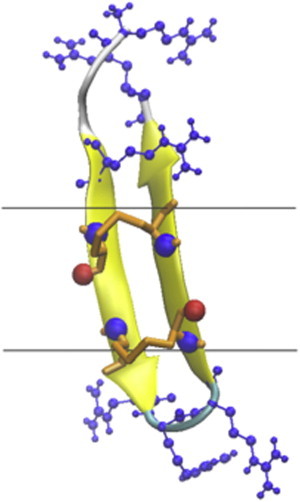
We considered both cylindrical and toroidal pores of different curvature. The radius of the pore was set to 13 Å and the membrane thickness to 26 Å. A PG1 monomer was initially placed perpendicular to the membrane at the pore interface at different starting positions, i.e., at the center of the pore and at the interface with either the SS side or the hydrophobic side facing the membrane interior. In each case, 1-ns simulations were run, and the average transfer energies to water were calculated over the last 0.6 ns. The peptide did not bind to the cylindrical pore, and most of it was out of the pore at the end of the simulations. In the toroidal pore, the binding energy depended on the curvature. No binding was observed for the low-curvature pore, whereas binding got stronger as the curvature increased (Table 3 reports the best of the three runs). Binding always occurred via the hydrophobic cluster. The structure of the peptide in the higher-curvature pores was tilted, especially in the k = 20 Å case (Fig. 4 shows the configuration with the most favorable transfer energy).
Table 3.
Transfer energies (kcal/mol) from water (w) and from flat membrane (m) to Ro = 13 Å pores in neutral, 30% anionic, and 100% anionic membranes
| w->neutral | w->30% an | w->100% an | m->30% an | m->100% an | |
|---|---|---|---|---|---|
| Cylindrical | −0.15 ± 0.3 | +0.9 ± 0.6 | −3.6 ± 0.4 | +4.8 ± 2.5 | +6.6 ± 1.2 |
| Toroidal k = 10 | −0.02 ± 0.03 | −9.1 ± 1.6 | −13.7 ± 1.7 | −3.9 ± 2.0 | −5.2 ± 1.7 |
| Toroidal k = 15 | −2.1 ± 0.6 | −8.8 ± 2.1 | −14.2 ± 2.5 | −4.6 ± 2.7 | −5.3 ± 4.0 |
| Toroidal k = 20 | −5.8 ± 0.4 | −9.6 ± 1.2 | −14.3 ± 2.5 | −4.4 ± 2.9 | −5.9 ± 1.9 |
Figure 4.
Cartoon representation in VMD of the structure of protegrin obtained in the simulations of the peptide in a neutral toroidal pore with R = 13 Å and k = 20 Å. The orange licorice representation indicates the position of the disulfide bonds, and the blue and silver sticks-and-balls represent the arginines and the hydrophobic cluster, respectively.
For anionic pores, we used a recently developed model with the electrostatic potential from numerical solution of the PB equation (Y. He, L. Prieto, and T. Lazaridis, unpublished). The results shown in Table 3 were obtained assuming a charge density at the center of the pore equal to 60% of that in the flat membrane. As expected, the transfer energy from water to the pore becomes more favorable as the membrane charge increases. The transfer energy from the flat membrane to the pore also becomes more favorable as the membrane charge increases. Transfer from the flat membrane to cylindrical pores is unfavorable. Transfer to toroidal pores is favorable and largely independent of curvature.
β-barrels in pores
Previous results from solid-state NMR suggested that protegrin forms β-barrels with parallel NCCN association of the peptides (23). However, in this model, the peptides point each of their sides in opposite directions, one toward the membrane and one toward the pore lumen. If one side is more favorable for membrane interaction than the other, one would expect that side to be always toward the membrane. Two other β-barrel models allow burial of the hydrophobic side of all peptides into the membrane: NCNC parallel and NCCN (same as CNNC) antiparallel. We constructed octameric models of these three types and tested their stability in implicit pores of radius 15 Å and varying curvature with 5-ns simulations.
As expected based on the monomer results, none of the three models were stable in cylindrical pores, and the peptides moved out of the pore within 1 ns. The barrels were much more stable in toroidal pores. Configurations of these models in optimal curvature pores at the end of the 5-ns simulation are shown in Fig. 5. The NCCN)par barrels collapse, forming essentially two interacting β-sheets with four peptides each. The NCCN)anti remain circular but some peptides tilt significantly. Most stable are the NCNC)par models, which retain a very regular structure. Table 4 shows the average effective energies and transfer energies for the toroidal pores. The parallel NCCN model has the highest average energy and unfavorable transfer energies from water to the pore. Apparently it is metastable in the pore, and in longer simulations it would move out. The other two models exhibit lower energies and favorable transfer energies to the pore. The NCNC)par model in higher-curvature pores exhibits the lowest average effective energy and also the most favorable transfer energy from water to the pore.
Figure 5.
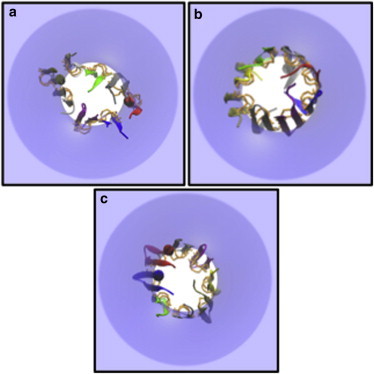
Top views of the minimized final conformations of protegrin octamers in membrane pores of Ro = 15 Å. (a) NCCN)par at k = 20 Å. (b) NCCN)anti at k = 15 Å. (c) NCNC)par at k = 20 Å. The disulfide bonds are highlighted by orange licorice.
Table 4.
Average effective energies and transfer energies (kcal/mol) from water to the pore of octameric barrels in toroidal pores from 5-ns simulations
| k = 10 Å | k = 15 Å | k = 20 Å | ||
|---|---|---|---|---|
| Neutral membrane | ||||
| NCCN)par | <W> | −3311 ± 19 | −3335 ± 10 | −3364 ± 13 |
| <ΔWtr> | +10.9 ± 1.0 | +11.2 ± 0.3 | +10.9 ± 0.6 | |
| NCCN)anti | <W> | −3366 ± 10 | −3378 ± 4 | −3374 ± 11 |
| <ΔWtr> | −5.8 ± 0.5 | −10.3 ± 0.9 | −12.8 ± 0.6 | |
| NCNC)par | <W> | −3388 ± 11 | −3393 ± 7 | −3395 ± 8 |
| <ΔWtr> | −19.1 ± 0.7 | −19.3 ± 1.0 | −20.4 ± 0.2 | |
| 30% Anionic membrane | ||||
| NCCN)par | <W> | −3567.7 ± 11.0 | −3563.1 ± 5.8 | −3578.6 ± 15.3 |
| <ΔWtr> | −37.6 ± 1.4 | −39.5 ± 4.1 | −37.7 ± 4.0 | |
| NCCN)anti | <W> | −3597.8 ± 2.6 | −3592.2 ± 7.2 | −3589.6 ± 4.7 |
| <ΔWtr> | −61.1 ± 1.1 | −59.9 ± 3.3 | −58.8 ± 2.8 | |
| NCNC)par | <W> | −3624.7 ± 8.4 | −3615.7 ± 7.8 | −3613.2 ± 7.5 |
| <ΔWtr> | −72.5 ± 0.8 | −69.0 ± 0.4 | −66.6 ± 1.0 | |
The same models were run in 30% anionic pores. Again, the NCCN)par arrangement is the least stable and exhibits the highest energies and the least favorable transfer energies (Table 4). The NCNC)par barrel exhibits the lowest effective energies and transfer energies to the pore. Fig. 6 shows the lowest-energy configuration of these octamers in anionic pores. NCNC)par and NCCN)anti are very stable, but the NCCN)par model collapses to a flat structure comprising two stacked β-sheets.
Figure 6.
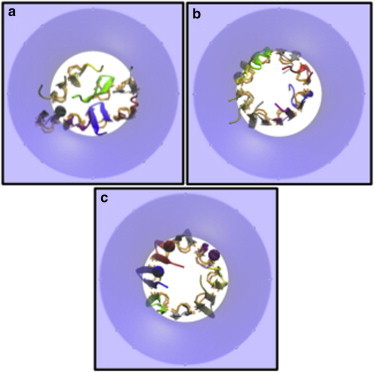
Top views of the lowest-energy conformation of protegrin octamer models in 30% anionic toroidal pores of Ro = 15 Å, k = 15 Å. (a) NCCN)par. (b) NCCN)anti. (c) NCNC)par. The latter has the lowest effective energy and the most favorable transfer energy to the membrane. The disulfide bonds are highlighted by orange licorice.
To check the implicit membrane results, we carried out 100-ns simulations in explicit POPC and POPE/POPG membranes (see Supporting Material). The NCNC)par barrel was found to be substantially more stable than the NCCN)par barrel in both membranes.
Pore formation energy
The energetic analysis above pertains to peptide binding to preformed pores. To determine whether a pore will form or not, one needs to add the free energy of pore formation/membrane deformation. This is a difficult undertaking. One could, however, make approximate estimates using experimental line tension values, γ. Typical values of the line tension range from 0.25 to 0.75 kT/Å (66). The free energy of forming a pore of radius R is 2πRγ. Therefore, for R = 15 Å and room temperature, the pore formation free energy will range from 14 to 42 kcal/mol. More recent experiments gave an average value of 7 pN for PC vesicles (67). With this value, the free energy of forming a 15 Å pore is 9.5 kcal/mol. The transfer energies of the NCNC)par barrel are barely larger than this value. It should be noted, however, that the transfer energies do not include any entropic effects, which are certainly unfavorable to pore formation. In any case, the range of values obtained suggests that pore formation in neutral membranes is energetically plausible, although perhaps somewhat difficult.
In anionic membranes, the relevant quantity is the difference in energy between the barrel and the peptides adsorbed on the flat membrane. If we take the adsorption energy of a monomer to the flat anionic membrane to be −4 kcal/mol, transfer of an octamer from the flat membrane to the pore would give about −83 – (−4) × 8 = −51 kcal/mol. Line tensions for anionic membranes are scarcer but they are of a similar order of magnitude. For example, a value of 6 pN was determined for dioleoylphosphatidylserine (68). Hence, we conclude that pore formation should be facile in anionic membranes. The above conclusions are consistent with the observed toxicity of protegrin toward red blood cells (which have membranes with a neutral outer leaflet) as well as their much higher activity in bacterial membranes (which have anionic membranes).
Discussion
The picture that emerges from these studies is that protegrin monomers interact very weakly with the surface of the zwitterionic membranes, but adsorb readily on the surface of anionic membranes. This result applies to intact bilayers and does not account for the possibility of membrane remodeling (69) or extraction of lipids to form peptide-lipid aggregates (70). Monomers do not insert in a TM orientation. Oligomers, however, could insert forming pores into both zwitterionic and anionic membranes. The most likely form of these oligomers is β-barrels, the most stable of which in our calculations has parallel NCNC topology. In these barrels, the hydrophobic cluster of protegrin is oriented toward the lipid and the SS bonds line the pore lumen. The presence of the hydrophobic cluster allows the lipids to bend only partially to form semitoroidal pores, as shown by explicit simulations here and in previous work (43,44). Combining the obtained effective energies with simple estimates of membrane deformation energy leads to the conclusion that pore formation should be facile in anionic bilayers but also possible in zwitterionic bilayers, consistent with the observed toxicity of protegrin toward mammalian cells.
We obtained the bulk of our results using an IMM. Such models are highly approximate. They account for hydrophobicity/hydrophilicity and generic protein-membrane electrostatic interactions, but not for specific lipid-protein interactions. They are also likely to have deficiencies in the treatment of intraprotein interactions, especially between arginines. On the other hand, they converge much faster than explicit simulations and are much less sensitive to initial conditions. As a partial check of the results of these calculations, we also performed explicit bilayer simulations of two β-barrels in two membranes (Supporting Material). The results seem consistent with the implicit membrane results; however, further explicit simulation tests, such as adsorption studies on zwitterionic membranes, would be helpful.
Our results challenge some aspects of the picture obtained by previous experimental and computational work. For example, the idea that monomers of protegrin insert into DLPC membranes (19) is not supported by our calculations. Several possible explanations can be offered: The solid-state NMR results (19,20) do not exclude the possibility that the PG1 peptides are associated with the membrane at membrane edges or pores, as suggested for DOPC. The fact that no loss of orientational order is observed for DLPC can be explained by the fact that the shorter DLPC chains do not need to reorient to form a semitoroidal pore. The critical experiment would be to show whether the S atoms interact with lipid or with water.
Furthermore, solid-state NMR revealed a parallel NCCN dimer in POPC membranes (22). We found that this type of dimerization is not favorable for interaction with either flat membranes or pores. Interestingly, we found that the NCCN)par dimer is slightly more stable than the others in aqueous solution, in agreement with experimental results for aqueous aggregates (65). Perhaps this intrinsic preference persists in the experimental samples, resulting in dimers that are primarily in the interstitial water layer interacting with two bilayers at the same time (71). It should be noted that the rotational-echo double-resonance (REDOR) experiments that established the NCCN)par topology (22), and the H and F spin diffusion experiments that showed contact of the peptide with the acyl chain region and oligomerization (23) were done under different conditions—the former at extended POPC bilayers at 35% hydration and the latter using ultracentrifuged large unilamellar vesicles.
In other experiments, Tang et al. (24) measured short distances between backbone and side-chain C atoms of Arg-4, Arg-11, Leu-5, and Val-16 with lipid phosphate atoms, supporting the idea of a toroidal pore. This result is clearly inconsistent with an NCNC)par barrel, in which all Arg-4 residues are facing the pore lumen (see Supporting Material). But it is also inconsistent with the NCCN)par barrel, in which half of the Arg-4 residues should face the pore lumen and should thus be far from lipid. The decay of the REDOR curves suggests that all peptides, not just a fraction, lie close to lipid headgroups. This is simply not possible in any barrel structure. Again, the only explanation we can think of is that these samples do not contain barrels, but rather dimers sandwiched between two different lipid bilayers. Crosspeaks between acyl chain protons and peptide 13C in H spin diffusion experiments (23,72) could also arise in the interfacial orientation.
Future studies should address explicitly the possibility of heterogeneity in the samples. The barrels are likely to be fragile structures, corresponding to a minor population in the samples. Previous work (22) considered only distances that are short in NCCN arrangements. Detection of a REDOR signal for these distances verifies the presence of NCCN dimers but does not exclude the possibility of a mixture of NCCN and NCNC dimers. It is important to also consider intermolecular distances that are short only in the NCNC arrangement, such as 15 C with 6 N. Appropriate isotopic labeling could distinguish between intermolecular and intramolecular contacts. Another possibility is to measure distances between atoms in the hydrophobic cluster side chains, which in the NCNC)par and NCCN)anti arrangements are on the same side of the hairpin and thus closer to each other.
Previous computational work focused on TM monomers and dimers (35,36,40,73) but did not examine the thermodynamic stability of these configurations. The peptides were initially placed in a TM configuration and may well have been kinetically stabilized by the large energy barrier of moving charges through the membrane. A very large, favorable insertion free energy was calculated for an anionic membrane (42) but not for PC membranes (Y. Kaznessis, University of Minnesota, personal communication, 2012). This is not consistent with any of the IMMs. It may well be that such models are fundamentally flawed, but at the same time we should carefully examine the explicit simulations for possible artifacts, such as bias arising from the starting structure, because bilayers are complex systems that relax very slowly. One might argue that the discrepancy is due to overestimation of the cost of Arg burial by IMMs (74), but, as shown above, it is backbone solvation and not Arg burial that is responsible for the unfavorable transfer energy in the thin membranes.
Octameric PG1 barrels have been simulated in explicit bilayers by two groups (43,44,46). One used the parallel NCCN configuration and the other used both parallel and antiparallel NCCN configurations. It is not clear which side was toward the membrane in the latter case. Our explicit simulations are not in conflict with these previous studies. We observed breaks in hydrogen bonding in the parallel NCCN barrel, as did Jang et al. (44), whereas Langham et al. (43) started with four dimers that were not hydrogen bonded to each other. The changes in the explicit simulations are not as dramatic as in our implicit simulations, which is likely due to the limited simulation time in the former; reconfiguration or movement out of the membrane should be a very slow process in explicit solvent. Ionic conductance calculations showed larger anion selectivity than was experimentally observed (47). The pore lumen has less positive charge in the NCCN)anti and NCNC)par barrels than in the NCCN)par barrel, and may give lower anion selectivity. Also, in the latter barrels, Arg-4 points toward the pore lumen. It is worth noting that protegrin 3, which has R4 mutated to G, is less anion selective than PG1 (6).
Heller et al. (27) detected two different states of protegrin in oriented lipid bilayers using oriented CD. They tentatively associated these states with a surface state and an inserted state, although they noted that the data were not consistent with a simple orientation change. Some irreversibility was observed, which may be a sign of kinetic trapping. The limitation of these experiments is that they do not reveal the oligomeric state of the peptide. Crystallization of protegrin pores was possible, but qualitative differences existed between protegrin and magainin pores (28).
The results presented here are consistent with recent atomic force microscopy experiments that showed that PG1 binds preferentially on bilayer edges at low peptide concentrations and reduces the line tension (75). At higher concentrations, the peptide began to bind to the lamellar regions and form pores of ∼9 nm diameter, which is much larger than the size inferred from polyethylene glycol blockage experiments (23). At even higher concentrations, the bilayer patches dissolved into worm-like micelles.
A lot remains for future work. First, the discrepancies between our results and previous experimental or computational work need to be resolved. Experiments on protegrin-membrane interactions in solution (e.g., using calorimetry, fluorescence, dye leakage, and giant unilamellar vesicle microscopy) would be invaluable in this regard. Another question relates to the optimal size of these aggregates. This could be determined by assessing the energetics of different oligomeric states and pore sizes using implicit membrane modeling. Ionic conductance calculations in all types of barrels will allow comparison with conductance and selectivity data. An important issue that we have not addressed here is the pathway of pore formation, especially for neutral membranes. How are these pore structures formed, what is the free-energy barrier, and what is the transition state? It is conceivable that local defects in the membrane are necessary for initiation of PG1 binding and pore formation.
Acknowledgments
We thank Prof. Ruth Stark for discussions about solid-state NMR.
This work was supported by the National Institutes of Health (SC1-AI084899) and utilized methods developed with the support of the National Science Foundation (MCB-0615552). Infrastructure support was provided in part by RCMI grant 2G12RR03060-26A1/8G12MD007603-27 from the National Institutes of Health. The CUNY High Performance Computing Center provided computational resources. Lidia Prieto received fellowships from the Ramón Areces Foundation and the Caja Madrid Foundation.
Supporting Material
References
- 1.Kokryakov V.N., Harwig S.S.L., Lehrer R.I. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993;327:231–236. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]
- 2.Fahrner R.L., Dieckmann T., Feigon J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996;3:543–550. doi: 10.1016/s1074-5521(96)90145-3. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg D.A., Hurst M.A., Fiddes J.C. Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997;41:1738–1742. doi: 10.1128/aac.41.8.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tam J.P., Wu C.W., Yang J.L. Membranolytic selectivity of cystine-stabilized cyclic protegrins. Eur. J. Biochem. 2000;267:3289–3300. doi: 10.1046/j.1432-1327.2000.01359.x. [DOI] [PubMed] [Google Scholar]
- 5.Mangoni M.E., Aumelas A., Chavanieu A. Change in membrane permeability induced by protegrin 1: implication of disulphide bridges for pore formation. FEBS Lett. 1996;383:93–98. doi: 10.1016/0014-5793(96)00236-0. [DOI] [PubMed] [Google Scholar]
- 6.Sokolov Y., Mirzabekov T., Kagan B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta. 1999;1420:23–29. doi: 10.1016/s0005-2736(99)00086-3. [DOI] [PubMed] [Google Scholar]
- 7.Chen J., Falla T.J., Fiddes J.C. Development of protegrins for the treatment and prevention of oral mucositis: structure-activity relationships of synthetic protegrin analogues. Biopolymers. 2000;55:88–98. doi: 10.1002/1097-0282(2000)55:1<88::AID-BIP80>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 8.Ostberg N., Kaznessis Y. Protegrin structure-activity relationships: using homology models of synthetic sequences to determine structural characteristics important for activity. Peptides. 2005;26:197–206. doi: 10.1016/j.peptides.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 9.Loury D.J., Embree J.R., Fiddes J.C. Effect of local application of the antimicrobial peptide IB-367 on the incidence and severity of oral mucositis in hamsters. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1999;87:544–551. doi: 10.1016/s1079-2104(99)70131-9. [DOI] [PubMed] [Google Scholar]
- 10.Elad S., Epstein J.B., Strahilevitz J. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J. Oral Pathol. Med. 2012;41:229–234. doi: 10.1111/j.1600-0714.2011.01094.x. [DOI] [PubMed] [Google Scholar]
- 11.Trotti A., Garden A., Ang K.K. A multinational, randomized phase III trial of iseganan HCl oral solution for reducing the severity of oral mucositis in patients receiving radiotherapy for head-and-neck malignancy. Int. J. Radiat. Oncol. Biol. Phys. 2004;58:674–681. doi: 10.1016/S0360-3016(03)01627-4. [DOI] [PubMed] [Google Scholar]
- 12.Robinson J.A., Shankaramma S.C., Obrecht D. Properties and structure-activity studies of cyclic β-hairpin peptidomimetics based on the cationic antimicrobial peptide protegrin I. Bioorg. Med. Chem. 2005;13:2055–2064. doi: 10.1016/j.bmc.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Harwig S.S.L., Waring A., Lehrer R.I. Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur. J. Biochem. 1996;240:352–357. doi: 10.1111/j.1432-1033.1996.0352h.x. [DOI] [PubMed] [Google Scholar]
- 14.Lai J.R., Huck B.R., Gellman S.H. Design of non-cysteine-containing antimicrobial β-hairpins: structure-activity relationship studies with linear protegrin-1 analogues. Biochemistry. 2002;41:12835–12842. doi: 10.1021/bi026127d. [DOI] [PubMed] [Google Scholar]
- 15.Lai J.R., Epand R.F., Gellman S.H. Roles of salt and conformation in the biological and physicochemical behavior of protegrin-1 and designed analogues: correlation of antimicrobial, hemolytic, and lipid bilayer-perturbing activities. Biochemistry. 2006;45:15718–15730. doi: 10.1021/bi0617759. [DOI] [PubMed] [Google Scholar]
- 16.Dawson R.M., Liu C.Q. Disulphide bonds of the peptide protegrin-1 are not essential for antimicrobial activity and haemolytic activity. Int. J. Antimicrob. Agents. 2010;36:579–580. doi: 10.1016/j.ijantimicag.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Hong M. Structure, topology, and dynamics of membrane peptides and proteins from solid-state NMR spectroscopy. J. Phys. Chem. B. 2007;111:10340–10351. doi: 10.1021/jp073652j. [DOI] [PubMed] [Google Scholar]
- 18.Tang M., Hong M. Structure and mechanism of β-hairpin antimicrobial peptides in lipid bilayers from solid-state NMR spectroscopy. Mol. Biosyst. 2009;5:317–322. doi: 10.1039/b820398a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaguchi S., Hong T., Hong M. Solid-state NMR investigations of peptide-lipid interaction and orientation of a β-sheet antimicrobial peptide, protegrin. Biochemistry. 2002;41:9852–9862. doi: 10.1021/bi0257991. [DOI] [PubMed] [Google Scholar]
- 20.Buffy J.J., Hong T., Hong M. Solid-state NMR investigation of the depth of insertion of protegrin-1 in lipid bilayers using paramagnetic Mn2+ Biophys. J. 2003;85:2363–2373. doi: 10.1016/s0006-3495(03)74660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buffy J.J., Waring A.J., Hong M. Determination of peptide oligomerization in lipid bilayers using 19F spin diffusion NMR. J. Am. Chem. Soc. 2005;127:4477–4483. doi: 10.1021/ja043621r. [DOI] [PubMed] [Google Scholar]
- 22.Mani R., Tang M., Hong M. Membrane-bound dimer structure of a β-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 2006;45:8341–8349. doi: 10.1021/bi060305b. [DOI] [PubMed] [Google Scholar]
- 23.Mani R., Cady S.D., Hong M. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA. 2006;103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang M., Waring A.J., Hong M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007;129:11438–11446. doi: 10.1021/ja072511s. [DOI] [PubMed] [Google Scholar]
- 25.Su Y.C., Waring A.J., Hong M. Structures of β-hairpin antimicrobial protegrin peptides in lipopolysaccharide membranes: mechanism of gram selectivity obtained from solid-state nuclear magnetic resonance. Biochemistry. 2011;50:2072–2083. doi: 10.1021/bi101975v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heller W.T., Waring A.J., Huang H.W. Multiple states of β-sheet peptide protegrin in lipid bilayers. Biochemistry. 1998;37:17331–17338. doi: 10.1021/bi981314q. [DOI] [PubMed] [Google Scholar]
- 27.Heller W.T., Waring A.J., Huang H.W. Membrane thinning effect of the β-sheet antimicrobial protegrin. Biochemistry. 2000;39:139–145. doi: 10.1021/bi991892m. [DOI] [PubMed] [Google Scholar]
- 28.Yang L., Weiss T.M., Huang H.W. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 2000;79:2002–2009. doi: 10.1016/S0006-3495(00)76448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gidalevitz D., Ishitsuka Y., Lee K.Y. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA. 2003;100:6302–6307. doi: 10.1073/pnas.0934731100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishitsuka Y., Pham D.S., Lee K.Y. Insertion selectivity of antimicrobial peptide protegrin-1 into lipid monolayers: effect of head group electrostatics and tail group packing. Biochim. Biophys. Acta. 2006;1758:1450–1460. doi: 10.1016/j.bbamem.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Bolintineanu D.S., Kaznessis Y.N. Computational studies of protegrin antimicrobial peptides: a review. Peptides. 2011;32:188–201. doi: 10.1016/j.peptides.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langham A.A., Kaznessis Y.N. Effects of mutations on the C-terminus of protegrin-1: a molecular dynamics simulation study. Mol. Simul. 2006;32:193–201. [Google Scholar]
- 33.Bolintineanu D.S., Langham A.A., Kaznessis Y.N. Molecular dynamics simulations of three protegrin-type antimicrobial peptides: interplay between charges at the termini, β-sheet structure and amphiphilic interactions. Mol. Simul. 2007;33:809–819. doi: 10.1080/08927020701393481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khandelia H., Kaznessis Y.N. Structure of the antimicrobial β-hairpin peptide protegrin-1 in a DLPC lipid bilayer investigated by molecular dynamics simulation. Biochim. Biophys. Acta. 2007;1768:509–520. doi: 10.1016/j.bbamem.2006.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandasamy S.K., Larson R.G. Binding modes of protegrin-1, a β-strand antimicrobial peptide, in lipid bilayers. Mol. Simul. 2007;33:799–807. [Google Scholar]
- 36.Rui H., Lee J., Im W. Comparative molecular dynamics simulation studies of protegrin-1 monomer and dimer in two different lipid bilayers. Biophys. J. 2009;97:787–795. doi: 10.1016/j.bpj.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang H., Ma B., Nussinov R. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J. 2006;91:2848–2859. doi: 10.1529/biophysj.106.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang H., Ma B.Y., Nussinov R. Conformational study of the protegrin-I (PG-I) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 2007;7:20. doi: 10.1186/1472-6807-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vivcharuk V., Kaznessis Y.N. Dimerization of protegrin-1 in different environments. Int. J. Mol. Sci. 2010;11:3177–3194. doi: 10.3390/ijms11093177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rui H.A., Im W. Protegrin-1 orientation and physicochemical properties in membrane bilayers studied by potential of mean force calculations. J. Comput. Chem. 2010;31:2859–2867. doi: 10.1002/jcc.21580. [DOI] [PubMed] [Google Scholar]
- 41.Vivcharuk V., Kaznessis Y. Free energy profile of the interaction between a monomer or a dimer of protegrin-1 in a specific binding orientation and a model lipid bilayer. J. Phys. Chem. B. 2010;114:2790–2797. doi: 10.1021/jp909640g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vivcharuk V., Kaznessis Y.N. Thermodynamic analysis of protegrin-1 insertion and permeation through a lipid bilayer. J. Phys. Chem. B. 2011;115:14704–14712. doi: 10.1021/jp205153y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langham A.A., Ahmad A.S., Kaznessis Y.N. On the nature of antimicrobial activity: a model for protegrin-1 pores. J. Am. Chem. Soc. 2008;130:4338–4346. doi: 10.1021/ja0780380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jang H., Ma B., Nussinov R. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic alzheimer β-amyloid ion channels. Biophys. J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jang H., Teran Arce F., Nussinov R. Structural convergence among diverse, toxic β-sheet ion channels. J. Phys. Chem. B. 2010;114:9445–9451. doi: 10.1021/jp104073k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capone R., Mustata M., Lal R. Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 2010;98:2644–2652. doi: 10.1016/j.bpj.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolintineanu D.S., Sayyed-Ahmad A., Kaznessis Y.N. Poisson-Nernst-Planck models of nonequilibrium ion electrodiffusion through a protegrin transmembrane pore. PLoS Comput. Biol. 2009;5:e1000277. doi: 10.1371/journal.pcbi.1000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mihajlovic M., Lazaridis T. Antimicrobial peptides bind more strongly to membrane pores. Biochim. Biophys. Acta. 2010;1798:1494–1502. doi: 10.1016/j.bbamem.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mihajlovic M., Lazaridis T. Charge distribution and imperfect amphipathicity affect pore formation by antimicrobial peptides. Biochim. Biophys. Acta. 2012;1818:1274–1283. doi: 10.1016/j.bbamem.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52:176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 51.Lazaridis T. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins. 2005;58:518–527. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- 52.Im W., Feig M., Brooks C.L., 3rd An implicit membrane generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazaridis T., Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 54.Mottamal M., Lazaridis T. Voltage-dependent energetics of alamethicin monomers in the membrane. Biophys. Chem. 2006;122:50–57. doi: 10.1016/j.bpc.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Zhan H., Lazaridis T. Influence of the membrane dipole potential on peptide binding to lipid bilayers. Biophys. Chem. 2012;161:1–7. doi: 10.1016/j.bpc.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lazaridis T. Structural determinants of transmembrane β-barrels. J. Chem. Theory Comput. 2005;1:716–722. doi: 10.1021/ct050055x. [DOI] [PubMed] [Google Scholar]
- 57.Reference deleted in proof.
- 58.Ben-Tal N., Ben-Shaul A., Honig B. Free-energy determinants of α-helix insertion into lipid bilayers. Biophys. J. 1996;70:1803–1812. doi: 10.1016/S0006-3495(96)79744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roux B. Influence of the membrane potential on the free energy of an intrinsic protein. Biophys. J. 1997;73:2980–2989. doi: 10.1016/S0006-3495(97)78327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi S., Takeshima K., Matsuzaki K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: proline as a translocation promoting factor. Biochemistry. 2000;39:8648–8654. doi: 10.1021/bi0004549. [DOI] [PubMed] [Google Scholar]
- 62.Fleming E., Maharaj N.P., Elmore D.E. Effect of lipid composition on buforin II structure and membrane entry. Proteins. 2008;73:480–491. doi: 10.1002/prot.22074. [DOI] [PubMed] [Google Scholar]
- 63.Aumelas A., Mangoni M., Chavanieu A. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 1996;237:575–583. doi: 10.1111/j.1432-1033.1996.0575p.x. [DOI] [PubMed] [Google Scholar]
- 64.Roumestand C., Louis V., Chavanieu A. Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett. 1998;421:263–267. doi: 10.1016/s0014-5793(97)01579-2. [DOI] [PubMed] [Google Scholar]
- 65.Tang M., Waring A.J., Hong M. Intermolecular packing and alignment in an ordered β-hairpin antimicrobial peptide aggregate from 2D solid-state NMR. J. Am. Chem. Soc. 2005;127:13919–13927. doi: 10.1021/ja0526665. [DOI] [PubMed] [Google Scholar]
- 66.May S. A molecular model for the line tension of lipid membranes. Eur. Phys. J. E. 2000;3:37–44. [Google Scholar]
- 67.Srividya N., Muralidharan S., Tripp B. Determination of the line tension of giant vesicles from pore-closing dynamics. J. Phys. Chem. B. 2008;112:7147–7152. doi: 10.1021/jp7119203. (Erratum in J. Phys. Chem. B. 2009. 113:7040) [DOI] [PubMed] [Google Scholar]
- 68.Loi S., Sun G., Butt H.J. Rupture of molecular thin films observed in atomic force microscopy. II. Experiment. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002;66:031602. doi: 10.1103/PhysRevE.66.031602. [DOI] [PubMed] [Google Scholar]
- 69.Lam K.L.H., Ishitsuka Y., Lee K.Y. Mechanism of supported membrane disruption by antimicrobial peptide protegrin-1. J. Phys. Chem. B. 2006;110:21282–21286. doi: 10.1021/jp0630065. [DOI] [PubMed] [Google Scholar]
- 70.Kim C., Wi S. A solid-state NMR study of the kinetics of the activity of an antimicrobial peptide, PG-1 on lipid membranes. Bull. Korean Chem. Soc. 2012;33:426–432. [Google Scholar]
- 71.Silvestro L., Axelsen P.H. Infrared spectroscopy of supported lipid monolayer, bilayer, and multibilayer membranes. Chem. Phys. Lipids. 1998;96:69–80. doi: 10.1016/s0009-3084(98)00081-4. [DOI] [PubMed] [Google Scholar]
- 72.Buffy J.J., Waring A.J., Hong M. Immobilization and aggregation of the antimicrobial peptide protegrin-1 in lipid bilayers investigated by solid-state NMR. Biochemistry. 2003;42:13725–13734. doi: 10.1021/bi035187w. [DOI] [PubMed] [Google Scholar]
- 73.Sayyed-Ahmad A., Kaznessis Y.N. Determining the orientation of protegrin-1 in DLPC bilayers using an implicit solvent-membrane model. PLoS ONE. 2009;4:e4799. doi: 10.1371/journal.pone.0004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hristova K., Wimley W.C. A look at arginine in membranes. J. Membr. Biol. 2011;239:49–56. doi: 10.1007/s00232-010-9323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lam K.L.H., Wang H., Lee K.Y. Mechanism of structural transformations induced by antimicrobial peptides in lipid membranes. Biochim. Biophys. Acta. 2012;1818:194–204. doi: 10.1016/j.bbamem.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 76.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.