

Abstract
To understand the variation of protein sequences in nature, we need to reckon with evolutionary constraints that are biophysical, cellular, and ecological. Here, we show that under the global selection against protein misfolding, there exists a scaling among protein folding stability, protein cellular abundance, and effective population size. The specific scaling implies that the several-orders-of-magnitude range of protein abundances in the cell should leave imprints on extant protein structures, a prediction that is supported by our structural analysis of the yeast proteome.
In molecular biophysics, the view that properties of proteins can be determined from first principles of physics and chemistry is almost a canon law. Advances in molecular dynamics, protein folding, ab initio structure prediction, and design of novel protein folds and function all support this view. Notwithstanding these developments, to what extent can physics and chemistry account for the diversity of biophysical and biochemical properties of proteins in nature?
From comparative genomics, one emerging constraint in the evolution of the coding regions of the genome is global selection against the cytotoxic effects of protein misfolding (1). Misfolded proteins are detrimental to the cell because they can form aggregates that can be toxic (2). The apparent universality of this constraint is manifested in the consistent observation that highly expressed proteins evolve more slowly across all forms of life—from bacteria to nematodes, mammals, and humans (1). Apart from explaining the universal correlation between abundance and the rate of evolution, a major prediction of the misfolding hypothesis is that more abundant proteins will evolve toward greater stability (1,3,4). One can show that this prediction arises from the interplay of population dynamics and protein biophysics.
Assuming monoclonality, the rate of protein evolution (ratio of nonsynonymous and synonymous substitutions) can be expressed as (5,6)
| (1) |
where Ne is the effective population size and s is the change in fitness due to the substitution (the selection coefficient). In a recent work (4), we showed that under the selection against protein misfolding, and assuming a two-state folding process, s is explicitly expressed as
| (2) |
where A is the cellular abundance of a protein, ΔG is the folding stability, c is the fitness cost per misfolded protein (measured in yeast to be ∼32/(total cellular protein concentration (7)), and β = 1/kBT. From Eq. 2, the rate is a function of premutation gene properties (abundance and ΔG) and the change in stability due to the arising mutation (ΔΔG).
Integrating over all possible mutational effects p(ΔΔG), the molecular clock surface is
| (3) |
The distribution p(ΔΔG) is approximately a Gaussian with mean ΔΔGmean (1 kcal/mol) and standard deviation ΔΔGsd (1.7 kcal/mol). Estimates for both parameters are derived from empirical measurements of folding stability changes due to single point mutations (ProTherm database (8)). This integral (Eq. 3) defines the molecular clock surface shown in Fig. 1. Because fixation of a mutation changes ΔG, the evolution of a gene is essentially a walk on the molecular clock surface, and this walk is slowest in the neighborhood of the gully (Fig. 1, red line). Consequently, on evolutionary timescales, genes tend to cluster in the gully of the surface under mutation-selection balance (4). Indeed, evolutionary simulations from various groups have predicted this correlation between abundance and stability (1,3,4).
Figure 1.

Rate of evolution of a protein as a function of its cellular abundance and folding stability. Rate is defined as dN/dS (the rate of nonsynymous substitutions per nonsynonymous sites). The rate is slowest in the gully (red line), which defines the average relationship among the folding stability, abundance, and population size (see Serohijos et al. (4) and Supporting Material).
The surface defined by Eq. 3 has a minimum at
| (4) |
According to the ProTherm database (8), most proteins have stabilities < −3 kcal/mol. In this regime, the above expression takes a simpler form:
| (5) |
or
| (6) |
which defines a peculiar scaling relationship among the average stability of proteins in a proteome (ΔG), their cellular abundance A, and the organism’s effective population size Ne. All of the variables on the right-hand side of Eq. 6 have been measured or estimated empirically, allowing one to assign the relative contribution of population size and abundance to the evolution of protein folding stability (Table 1). Indeed, the variation of protein folding stability in nature could be largely due to protein abundance and population size (Table 1).
Table 1.
Energetic equivalence of constraints imposed by evolutionary variables
| Variablea | Observed/estimated values in nature | Energetic equivalence (kcal/mol) |
|---|---|---|
| A | 10–106 (9) | −1 to −8 |
| Ne | 104–108 (17) | −5 to −11 |
We used the scaling in Eq. 6 to assign the relative strength of constraints imposed by the evolutionary variables on the evolution of folding stability (ΔG). Abundance has been measured in yeast. Effective population sizes have been estimated across all kingdoms of life (104 in mammals and 108 in prokaryotes). The calculation assumes monoclonality (μ ≪ 1), and the effect of the mutation rate μ on the scaling remains an open question. kBT = 0.593 kcal/mol.
Considering that protein cellular abundances span 10–106 copies per cell (as shown in yeast (9)), with an energetic equivalence of ∼7 kcal/mol in protein stability (Table 1), we reasoned that abundance should systematically manifest in the structural properties of proteins across a genome. To date, the strongest empirical support for the interdependence of abundance and stability is the observation that highly abundant, slowly evolving proteins and proteins from thermophilic bacteria share a similar amino acid composition (10). To demonstrate this prediction more unambiguously, we extracted all of the yeast proteins from the Protein Data Bank, partitioned them into domains as defined by SCOP (11), and then mapped their experimentally measured abundance (9). Also, we excluded domains with gaps in the structure. This procedure yielded 302 domains on which we performed a structural analysis (Fig. 2 and Table S1 in the Supporting Material). Using the modeling tool Eris (12), we calculated the hydrogen-bonding energy and van der Waals interaction energy (two major contributors to the folding free energy) within each domain (13). Residues in more abundant proteins form more extensive hydrogen bonds between their side chains and backbones (r = −0.29∗∗∗) and among their side chains (r = −0.30∗∗∗). Abundance likewise correlates with increasing van der Waals interaction (r = −0.30∗∗∗).
Figure 2.
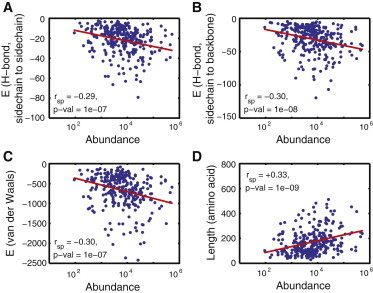
(A–C) Correlation between abundance and structural properties (hydrogen-bond content and strength of van der Waals interaction) of protein domains in yeast. (D) Stability is an extensive property, and thus abundance correlates with domain length (14). Indicated are the values of the Spearman rank correlation. See also Table S1.
We note, however, that the manisfestation of the scaling (Eq. 6) is strong on protein structural properties that directly influence stability (Fig. 2, A–C), but could be less manifested in indirect indictators of stability, such as protein length (14). For example, in the 302 proteins we analyzed, the more-abundant domains were generally longer (r = 0.33∗∗∗). When we expanded the set to include domains (15) that do not have empirically determined structures (Fig. S1), we found no correlation between domain length and abundance, because length is a coarse descriptor of stablity. The general observation that more abundant genes tend to be shorter (r = −0.19∗∗∗) reflects the fact that they have fewer domains (r = −0.12∗∗∗; Fig. S1).
As was recently pointed out (16), population size constrains the cellular distribution of folding stabilities such that organisms with small effective population sizes (e.g., endosymbiotic parasites that undergo episodic bottlenecking) will evolve less thermodynamically stable proteins, simply because deleterious mutations will fix at a higher probability in smaller population sizes. On the contrary, organisms with higher population sizes, which experience stronger purifying selection, are predicted to evolve more stable proteins. Additionally, assuming that all other things are equal, vertebrates (with effective population sizes of 104–105 (17)) are predicted by Eq. 6 to evolve proteins that are on average 6 kcal/mol less stable than proteins in prokaryotes (whose population sizes are ≥108 (17); Table 1). Systematically proving this prediction is the subject of future work. Nonetheless, protein structures of viruses, which undergo episodic bottlenecking (and hence have a low effective population size), already show low van der Waals and hydrogen-bond contact densities (18).
Stability is the most universal and well understood biophysical property of proteins, and successes in protein folding and engineering are testaments to how much we understand stability from first principles. However, in nature, protein evolution must reckon with the stochastic processes of mutation and purifying selection, making the effective population size a crucial variable (16). Protein evolution likewise needs to reckon with emerging constraints in cell biology, such as the selection against protein misfolding (1,19), where abundance scales with the selective pressure felt by an evolving gene.
Acknowledgments
We thank N. Dokholyan for the use of ERIS, and Z. Rimas for discussions.
This work was supported by the National Institutes of Health. S.Y.R. Lee received funding from the Harvard College Research Program.
Supporting Material
References and Footnotes
- 1.Drummond D.A., Wilke C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 3.Yang J.R., Zhuang S.M., Zhang J. Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol. Syst. Biol. 2010;6:421. doi: 10.1038/msb.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serohijos A.W., Rimas Z., Shakhnovich E.I. Protein biophysics explains why highly abundant proteins evolve slowly. Cell Rep. 2012;2:249–256. doi: 10.1016/j.celrep.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Z., Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 6.Kryazhimskiy S., Plotkin J.B. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiler-Samerotte K.A., Dion M.F., Drummond D.A. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc. Natl. Acad. Sci. USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar M.D., Bava K.A., Sarai A. ProTherm and ProNIT: thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 2006;34(Database issue):D204–D206. doi: 10.1093/nar/gkj103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghaemmaghami S., Huh W.K., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 10.Cherry J.L. Highly expressed and slowly evolving proteins share compositional properties with thermophilic proteins. Mol. Biol. Evol. 2010;27:735–741. doi: 10.1093/molbev/msp270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murzin A.G., Brenner S.E., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 12.Yin S., Ding F., Dokholyan N.V. Eris: an automated estimator of protein stability. Nat. Methods. 2007;4:466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 13.Kortemme T., Morozov A.V., Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh K., Dill K. Cellular proteomes have broad distributions of protein stability. Biophys. J. 2010;99:3996–4002. doi: 10.1016/j.bpj.2010.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malmström L., Riffle M., Baker D. Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol. 2007;5:e76. doi: 10.1371/journal.pbio.0050076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wylie C.S., Shakhnovich E.I. A biophysical protein folding model accounts for most mutational fitness effects in viruses. Proc. Natl. Acad. Sci. USA. 2011;108:9916–9921. doi: 10.1073/pnas.1017572108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynch M., Conery J.S. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 18.Tokuriki N., Oldfield C.J., Tawfik D.S. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009;34:53–59. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y., Dokholyan N.V. Natural selection against protein aggregation on self-interacting and essential proteins in yeast, fly, and worm. Mol. Biol. Evol. 2008;25:1530–1533. doi: 10.1093/molbev/msn122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.